
4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリンの製造方法
4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリン、その塩の製造方法、およびその製造に用いる中間体を記載する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバメトイルミル)ピペリジン−4−イル]オキシ}キナゾリンまたはその塩(以下、「化合物(I)」)の製造方法、および化合物(I)の製造に用いる中間体に関する。
【背景技術】
【0002】
式(I)の化合物は、国際特許出願公開番号WO2005/028469に例1として開示され、以
下の構造である。
【0003】
【化1】

【0004】
化合物(I)はerbB受容体型チロシンキナーゼの阻害薬であり、特に化合物(I)はEGFRおよびerbB2受容体型チロシンキナーゼの有効な阻害薬である。化合物(I)は、リガンド刺激されたEGFR/erbB3および/またはerbB2/erbB3ヘテロ二量体化に続くerbB3リン酸化の阻害により、erbB3仲介シグナル伝達をも阻害する。化合物(I)は、癌などの過増殖性障害の処置に有用であると予想される。
【0005】
WO 03/082831には、種々の4−(3−クロロ−2−フルオロアニリノ)キナゾリン類の製造が開示されている。しかし、化合物(I)はそこに開示されていない。
【0006】
WO2005/028469には、例1として化合物(I)の製造法が以下のように開示されている
。
【0007】
2−クロロ−N−メチルアセトアミド(32mg,0.3mmol)を、4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−[(ピペリジン−4−イル)オキシ]キナゾリン(120mg,0.3mmol)、ヨウ化カリウム(16mg,0.1mmol)および炭酸カリウム(50mg,0.36mmol)のアセトニトリル(5ml)中における混合物に添加した。この混合物を1時間、加熱還流した。溶媒を真空下で蒸発させた後、残留物をジクロロメタンに装入した。この有機溶液を水およびブラインで洗浄し、硫酸マグネシウムで乾燥させた。溶媒を真空下で蒸発させた後、残留物をシリカゲル上でのクロマトグラフィー(溶離剤:ジクロロメタン中、1%−2%の7Nメタノール性アンモニア)により精製して、化合物(I)が得られた。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】WO 2005/028469
【特許文献2】WO 03/082831
【発明の概要】
【0009】
本発明者らは化合物(I)を製造するための別法を見出した。この方法は、少ない数の製造工程で高収率において不純物が最少である化合物(I)を提供する。したがってこの方法は、化合物(I)の大規模製造に使用するのに適している。
【0010】
1つの態様は、4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリンまたはその塩の製造方法であって:
(a)式(II)の化合物:
【0011】
【化2】
【0012】
を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させること;または
(b)式(III)の化合物:
【0013】
【化3】
【0014】
を適切な酸の存在下で式(XI)または式(XII)の化合物:
【0015】
【化4】
【0016】
と反応させること;
を含む方法を提供する。
【図面の簡単な説明】
【0017】
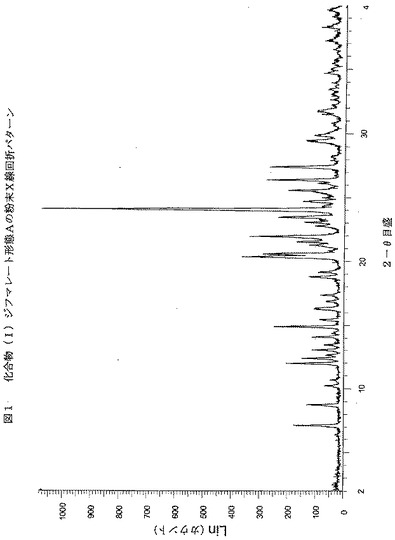
【図1】図1は、化合物(I)ジフマレート形態AについてのX線粉末回折パターン(XRPD)を示す。x軸は2θ値を示し、y軸はカウントを示す。
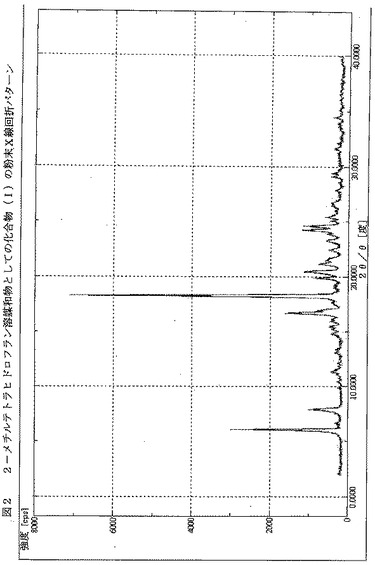
【図2】図2は、2−メチルテトラヒドロフラン溶媒和物としての化合物(I)のX線粉末回折パターン(XRPD)を示す。x軸は2θ値を示し、y軸はカウントを示す。
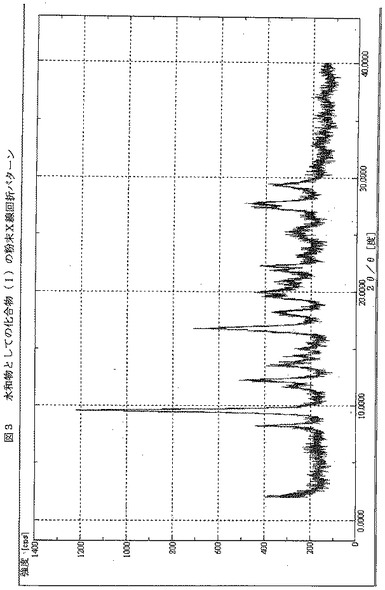
【図3】図3は、水和物としての化合物(I)のX線粉末回折パターン(XRPD)を示す。x軸は2θ値を示し、y軸はカウントを示す。
【図4】図4は、イソプロパノール溶媒和物としての化合物(I)のX線粉末回折パターン(XRPD)を示す。x軸は2θ値を示し、y軸はカウントを示す。
【発明を実施するための形態】
【0018】
1態様は、4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリンまたはその塩の製造方法であって:
(a)式(II)の化合物:
【0019】
【化5】
【0020】
を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させることを含む方法を提供する。
【0021】
この反応は、適切には、適切な酸、たとえば酢酸、ブタン二酸、プロパン酸、コハク酸、フマル酸およびクエン酸、またはその混合物から選択される1種類以上の酸の存在下で実施される。本発明の1態様において、酸は酢酸である。
【0022】
この反応は、適切には、不活性溶媒、たとえばシクロヘキサン、芳香族炭化水素系溶媒、たとえばトルエン、メトキシベンゼンもしくはキシレン;ニトリル系溶媒、たとえばアセトニトリル;エーテル類、たとえば2−メチルテトラヒドロフラン;またはエステル、たとえば酢酸イソプロピルの存在下で実施される。1態様において、溶媒はトルエン、シクロヘキサン、メトキシベンゼン、キシレン、アセトニトリル、2−メチルテトラヒドロフランおよび酢酸イソプロピルから選択される。他の態様において、溶媒はトルエン、シクロヘキサン、メトキシベンゼンおよびキシレンから選択される。さらに他の態様において、溶媒はメトキシベンゼンである。
【0023】
この反応は、適切には高められた温度、たとえば約80から約120℃まで、たとえば約90〜120℃、適切には約90℃で実施される。
【0024】
適切には、式(II)の化合物に対して等モルまたは過剰モルの3−クロロ−2−フルオロアニリンを使用する。たとえば、3−クロロ−2−フルオロアニリンと式(II)の化合物のモル比は、約1:1から約1:2まで、適切には約1:1である。
【0025】
いずれの態様においても、化合物(I)を常法により単離することができる。たとえば、実施例に記載するように化合物(I)を水中に抽出し、そして溶液から結晶化させることができる。必要であれば、溶液からの化合物(I)の結晶化はその溶液に化合物(I)の種晶を添加することにより開始できる。生成した固体を次いで常法により、たとえば化合物(I)の濾過および乾燥により採集することができる。
【0026】
本発明のさらに他の観点によれば、式(II)の化合物の製造方法であって、式(III)の化合物:
【0027】
【化6】
【0028】
をN,N−ジメチルホルムアミドジメチルアセタールと反応させることを含む方法が提供される。
【0029】
この反応は、適切には酸性条件下で実施される。たとえば、この反応は適切には酢酸の存在下で実施される。
【0030】
この反応は、好都合には適切な溶媒、たとえばエーテル類、たとえば2−メチルテトラヒドロフラン、または芳香族炭化水素、たとえばトルエンの存在下で実施される。この反応は、適切には高められた温度、たとえば約70〜105℃、適切には約76℃で実施される。
【0031】
他の態様において、化合物(I)は、式(III)の化合物をN,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミド(化合物(XI))と反応させることにより、式(III)の化合物から直接製造できる。この反応は、適切には酸性条件下で実施される。この反応は、適切には、適切な酸、たとえば酢酸、ブタン二酸、プロパン酸、コハク酸、フマル酸およびクエン酸、またはその混合物から選択される1種類以上の酸の存在下で実施される。本発明の1態様において、酸はフマル酸である。
【0032】
この反応は、好都合には適切な溶媒、たとえばエーテル類、たとえば2−メチルテトラヒドロフランの存在下で実施される。この反応は、適切には高められた温度、たとえば約70〜105℃、適切には約90℃で実施される。
【0033】
他の態様において、化合物(I)は、式(III)の化合物をN’−(3−クロロ−2−フルオロ−フェニル)−N,N−ジメチル−ホルムアミジン(化合物(XII))と反応させることにより、式(III)の化合物から直接製造できる。この反応は、適切には酸性条件下で実施される。この反応は、適切には、適切な酸、たとえば酢酸、ブタン二酸、プロパン酸、コハク酸、フマル酸およびクエン酸、またはその混合物から選択される1種類以上の酸の存在下で実施される。本発明の1態様において、酸はフマル酸である。
【0034】
式(III)の化合物は、式(IV)の化合物:
【0035】
【化7】
【0036】
の還元を含む方法により製造できる。
【0037】
ニトロ基をアミンに還元するのに適した還元反応は周知である。たとえば、式(IV)の化合物を、適切な還元剤、たとえば亜ジオチン酸ナトリウムの存在下での還元により還元することができる。この反応は適切には水性溶媒、たとえば水性メタノールの存在下で実施される。この反応は、好都合には高められた温度、たとえば40〜60℃で実施される。
【0038】
あるいは、式(IV)の化合物の還元は水素化により、たとえば適切な触媒、たとえばカーボン上パラジウム触媒、たとえばカーボン上10%パラジウム触媒、または白金/バナジウム触媒、たとえばカーボン上1%白金+2%バナジウム触媒を用いる接触水素化により実施できる。この水素化は好都合には水性溶媒、たとえばメタノールまたはアセトニトリルの存在下で実施される。他の態様において、別の溶媒、たとえばメタノール、イソプロパノール、または1:1の比率のメタノール:イソプロパノールの混合物も使用できる。
【0039】
式(IV)の化合物は、式(V)の化合物:
【0040】
【化8】
【0041】
のニトロ化を含む方法により製造できる。
【0042】
式(V)の化合物のニトロ化は、芳香環のニトロ化のための周知の方法を用いて、たとえばそのような反応について周知の条件を用いて本明細書の実施例に示すように、式(V)の化合物を硫酸の存在下に硝酸で処理することにより実施できる。
【0043】
式(V)の化合物は、式(VI)の化合物:
【0044】
【化9】
【0045】
を式(VII)の化合物:
【0046】
【化10】
【0047】
(式中、Lg1は適切な脱離基である)と反応させることを含む方法により製造できる。
【0048】
Lg1により表わされる適切な脱離基には、たとえばハロゲノ、たとえばクロロが含まれる。
【0049】
この反応は、適切には、適切な塩基、たとえばカーボネート、有機アミンまたはアルコキシドの存在下で実施される。具体的な塩基には、たとえば炭酸カリウムまたはトリエタノールアミンが含まれる。
【0050】
この反応は、好都合には、不活性溶媒、たとえばアセトニトリル、またはアルコール類、たとえばエタノールの存在下で実施される。この反応は、適切には、高められた温度、好都合には溶媒の還流温度で実施される。
【0051】
式(VI)の化合物は、たとえば反応スキーム1に示すようにして製造できる:
【0052】
【化11】
【0053】
(スキーム1の脚注)
工程(i):Lg2は適切な脱離基、たとえばハロゲノ、アルカンスルホニルオキシまたはアリールスルホニルオキシ基、たとえばクロロ、ブロモ、メタンスルホニルオキシ、4−ニトロベンゼンスルホニルオキシまたはトルエン−4−スルホニルオキシ基である。適切にはLg2はメタンスルホニルオキシ、4−ニトロベンゼンスルホニルオキシまたはトルエン−4−スルホニルオキシ基であり、たとえばLg2はメタンスルホニルオキシである。
【0054】
Pg1は適切なアミン保護基である。そのような基は、たとえばこの主題に関する多数の一般的テキストのいずれかに記載されているように周知である:たとえば‘Protective
Groups in Organic Synthesis’, Theodora Green著 (出版社: John Wiley & Sons)。アミノ保護基の例には、アシル基、たとえばアルカノイル基、たとえばアセチル、アルコキシカルボニル基、たとえばメトキシカルボニル、エトキシカルボニルもしくはtert−ブトキシカルボニル基、アリールメトキシカルボニル基、たとえばベンジルオキシカルボニル、またはアロイル基、たとえばベンゾイルが含まれる。Pg1の格別な例はtert−ブトキシカルボニルである。
【0055】
この反応は、適切には塩基、たとえばカーボネート、たとえば炭酸カリウムの存在下で実施される。この反応は、好都合には、適切な不活性溶媒、たとえばアルコール類、たとえばイソプロパノールの存在下で実施される。この反応は、適切には、高められた温度、好都合には溶媒の還流温度で実施される。
【0056】
工程(ii):保護基Pg1は常法により除去される。たとえばPg1がtert−ブ
トキシカルボニルである場合、それは塩酸、硫酸もしくはリン酸またはトリフルオロ酢酸など適切な酸で処理することにより除去できる。
【0057】
他の態様において、化合物(I)は、式(III)の化合物を式(XI)の化合物と反応させることにより、式(III)の化合物から直接製造できる。
【0058】
【化12】
【0059】
式(XI)の化合物はN,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミドとも呼ばれる。他の態様において、化合物(I)は、式(III)の化合物を式(XII)の化合物と反応させることにより、式(III)の化合物から直接製造できる。
【0060】
【化13】
【0061】
式(XII)の化合物はN’−(3−クロロ−2−フルオロ−フェニル)−N,N−ジメチル−ホルムアミジンと呼ばれる。これらの反応はいずれも、適切には酸性条件下で実施される。たとえば、この反応は、適切には酢酸、ブタン二酸、フマル酸またはプロパン酸の存在下で実施される。
【0062】
この反応は、好都合には適切な溶媒、たとえばエーテル類、たとえば2−メチルテトラヒドロフラン、またはアルコール類、たとえばエタノールまたはtert−ブチルアルコールの存在下で実施される。この反応は、適切には高められた温度、たとえば約70〜約105℃で実施される。ある態様において、この反応は約80〜約90℃で実施できる。
【0063】
式(XI)の化合物は、3−クロロ−2−フルオロアニリンをオルトギ酸エチルと反応させることにより製造できる。この反応は、適切には酸性条件下で実施される。たとえば、この反応は、適切には酢酸の存在下で実施される。
【0064】
この反応は、好都合には適切な溶媒、たとえばシクロヘキサンの存在下で実施される。この反応は、適切には高められた温度、たとえば約40〜約60℃、適切には約50℃で実施される。
【0065】
したがって、化合物(I)は式(III)の化合物から前記のいずれかの経路で製造できる。
【0066】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0067】
工程1〜2に適切な条件は前記に定めたとおりである。
【0068】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(3a)前記に定めた式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程。
【0069】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(2b)前記に定めた式(III)の化合物を前記に定めた式(XI)の化合物と反応させる工程;および
(1)化合物(I)を単離する工程。
【0070】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(3b)3−クロロ−2−フルオロアニリンをオルトギ酸エチルと反応させて、式(XI)の化合物にする工程。
【0071】
あるいは、本発明の他の観点において、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(2c)前記に定めた式(III)の化合物を前記に定めた式(XII)の化合物と反応させる工程;および
(1)化合物(I)を単離する工程。
【0072】
上記方法はさらに下記の工程を含むことができる:
(3c)3−クロロ−2−フルオロアニリンをN,N−ジメチルホルムアミドジメチルアセタールと反応させて、式(XII)の化合物にする工程。
【0073】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(4)前記に定めた式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程。
【0074】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(5)前記に定めた式(V)の化合物をニトロ化して、前記に定めた式(IV)の化合物にする工程。
【0075】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(6)前記に定めた式(VI)の化合物を前記に定めた式(VII)の化合物と反応させて、前記に定めた式(V)の化合物にする工程。
【0076】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(7)式(VIII)の化合物を脱保護して、前記に定めた式(VI)の化合物にする工程。
【0077】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(8)前記に定めた式(X)の化合物を前記に定めた式(IX)の化合物と反応させて、前記に定めた式(VIII)の化合物にする工程。
【0078】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(3a)前記に定めた式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0079】
工程1〜3に適切な条件は前記に定めたとおりである。
【0080】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(4)前記に定めた式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程;
(3a)式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0081】
工程1〜4に適切な条件は前記に定めたとおりである。
【0082】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(5)前記に定めた式(V)の化合物をニトロ化して、前記に定めた式(IV)の化合物にする工程;
(4)式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程;(3a)式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;そして
(1)化合物(I)を単離する工程。
【0083】
工程1〜5に適切な条件は前記に定めたとおりである。
【0084】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(6)前記に定めた式(VI)の化合物を前記に定めた式(VII)の化合物と反応させて、前記に定めた式(V)の化合物にする工程;
(5)式(V)の化合物をニトロ化して、前記に定めた式(IV)の化合物にする工程;(4)式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程;(3a)式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0085】
工程1〜6に適切な条件は前記に定めたとおりである。
【0086】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(8)前記に定めた式(X)の化合物を前記に定めた式(IX)の化合物と反応させて、
前記に定めた式(VIII)の化合物にする工程;
(7)式(VIII)の化合物を脱保護して、前記に定めた式(VI)の化合物にする工程;
(6)式(VI)の化合物を前記に定めた式(VII)の化合物と反応させて、前記に定めた式(V)の化合物にする工程;
(5)式(V)の化合物をニトロ化して、前記に定めた式(IV)の化合物にする工程;(4)式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程;(3a)式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0087】
工程1〜8に適切な条件は前記に定めたとおりである。
【0088】
前記のいずれかの方法において、工程2a)および3a)を下記の工程2b)および3b)で置き換えることができる;
(2b)前記に定めた式(III)の化合物を前記に定めた式(XI)もしくは(XII)の化合物と反応させる工程;および/または
(3b)3−クロロ−2−フルオロアニリンをオルトギ酸エチルと反応させて、式(XI)の化合物にする工程。
【0089】
前記のいずれかの方法において、工程2a)および3a)を下記の工程2c)および3c)で置き換えることができる;
(2c)前記に定めた式(III)の化合物を前記に定めた式(XII)の化合物と反応させる工程;および/または
(3c)3−クロロ−2−フルオロアニリンをN,N−ジメチルホルムアミドジメチルアセタールと反応させて、式(XII)の化合物にする工程。
【0090】
所望により、目的化合物(I)を医薬的に許容できる塩に変換することができる。WO2005/028469に化合物(I)の塩類の例、たとえば化合物(I)と無機酸または有機酸、た
とえば塩酸、臭化水素酸、硫酸、トリフルオロ酢酸、クエン酸またはマレイン酸との酸付加塩が記載されている。格別な塩は実施例に記載するように化合物(I)のジフマレート塩(二フマル酸塩)である。
【0091】
本発明による方法に使用する特定の中間体は新規であり、本発明のさらに他の観点をなす。本発明において提供される中間体またはその塩は下記の構造をもつことができる:
【0092】
【化14】
【0093】
式中:
R1は、H、−NH2、−NO2、または
【0094】
【化15】
【0095】
であり;
R2は、H、N−メチルカルバモイルメチル、またはPg1であり;
Pg1は、アミノ保護基であり;
ただし、R2がHまたはアミノ保護基である場合はR1はHである。
【0096】
したがって、本発明の他の観点は、式(II)、(III)、(IV)、(V)、(VI)および(VIII)のいずれかの化合物、またはその塩から選択される化合物を提供する。本発明の他の観点は、式(XI)の化合物またはその塩を提供する。本発明の他の観点は、式(XII)の化合物またはその塩を提供する。
【0097】
1態様において、たとえば式(VIII)の化合物においてPg1がtert−ブトキシカルボニルである化合物が提供される。
【0098】
式(II)、(XI)および(XII)の化合物を含めて本明細書に記載する化合物は幾何異性中心をもつ場合があり、E−およびZ−異性体として存在する可能性がある。本発明はそのような幾何異性体およびその混合物をすべて包含すると理解すべきである。本発明の1態様において、式(II)の化合物は実質的にE−異性体として存在する。本発明の他の態様において、式(II)の化合物は実質的にZ−異性体として存在する。本発明の1態様において、式(XI)の化合物は実質的にE−異性体として存在する。本発明の他の態様において、式(XI)の化合物は実質的にZ−異性体として存在する。本発明の1態様において、式(XII)の化合物は実質的にE−異性体として存在する。本発明の他の態様において、式(XII)の化合物は実質的にZ−異性体として存在する。
【0099】
これらの中間体は遊離塩基として、または適切な塩の形で使用できる。そのような塩類には、医薬的に許容できる塩類および医薬的に許容できない塩類の両方が含まれる。医薬的に許容できない塩の形の中間体の使用は、本発明による方法に有利な場合がある。たとえば、そのような塩類は中間体の単離または精製のために有用な可能性がある。必要であれば、それらの中間体を常法により修飾して、その化合物の医薬的に許容できる塩にすることができる。そのような方法は当業者に周知であり、たとえば医薬的に許容できる対イオンの存在下での化合物のイオン交換法または再沈殿を含む。したがって、本発明は前記の中間体およびその塩類を含むものであると理解すべきである。
【0100】
化合物(I)およびその塩類、たとえばジフマレート塩は、適切には適切な医薬組成物、たとえば錠剤、カプセル剤または顆粒配合物の形で患者に経口投与される。
【0101】
たとえば化合物(I)のジフマレートは、適切には下記の賦形剤を用いて錠剤として配合される:
錠剤コア:
化合物(I)のジフマレート(たとえば形態A);
乳糖;
微結晶性セルロース;
クロスポビドン;
ポリビドン(PVP);および
ステアリン酸マグネシウム。
【0102】
錠剤コアをフィルムコーティング、たとえばHPMCベースのフィルムコーティングでコートすることができ、このコーティングは場合により1種類以上の着色剤および/または光保護剤を含有する。
【0103】
錠剤は常法により、実施例に示したように製造できる。
【0104】
1種類以上の賦形剤と合わせて単一剤形を製造する有効成分の量は、処置されるホストおよび個々の投与経路に応じて必然的に異なるであろう。たとえば、ヒトに経口投与するための配合物は一般に、たとえば0.5mgから0.5gまでの有効薬剤(より適切には、0.5から200mgまで、たとえば1から100mgまで)を、適切かつ好都合な量の賦形剤と配合して含有するであろう;賦形剤は組成物全体の約5から約98重量%の範囲であってもよい。
【0105】
治療または予防のための化合物(I)およびその塩類、たとえばジフマレート塩の用量サイズは、その状態の性質および重症度、動物または患者の年齢および性別、ならびに投与経路に従い、周知の医薬原理に従って、当然異なるであろう。1態様において、癌、たとえば乳癌の処置に用いるのに適切な化合物(I)の量は、40、80、100、160、200または240mgを1日2回である。
【0106】
化合物(I)は抗増殖特性、たとえば抗癌特性をもち、これはそれらのerbBファミリー受容体型チロシンキナーゼ阻害活性、特に混合erbB2/EGFおよび/またはerbB3/EGFプロフィールから生じると考えられる。
【0107】
したがって、本発明の化合物は、もっぱらまたは部分的にerbB受容体型チロシンキナーゼにより仲介される疾患または病的状態の処置に有用であると予想される。したがって、本発明の化合物は、乾癬、良性前立腺肥大(BPH)、アテローム性硬化症および再狭窄および/または癌の処置に際して、抗増殖作用をもたらすことにより、特にerbB受容体型チロシンキナーゼ感受性癌の処置に有用であると予想される。そのような良性または悪性腫瘍はいかなる組織にも影響を及ぼす可能性があり、これには非充実性腫瘍、たとえば白血病、多発性骨髄腫またはリンパ腫、ならびに充実性腫瘍、たとえば胆管癌、骨癌、膀胱癌、脳/CNS癌、乳癌、結腸直腸癌、子宮内膜癌、胃癌、頭頚部癌、肝癌、肺癌、神経癌、食道癌、卵巣癌、膵臓癌、前立腺癌、腎癌、皮膚癌、精巣癌、甲状腺癌、子宮癌および外陰癌が含まれる。特に、化合物(I)は乳癌の処置に有用であると予想される。
【0108】
化合物(I)およびその塩類、たとえばジフマレートは、エストロゲンおよび/またはプロゲステロン陽性乳癌の処置に際して、有効量のアロマターゼ阻害薬、たとえばアナストロゾール(anastrozole)と組み合わせて使用できる。この組合わせは、以前に内分泌
療法、たとえば選択的エストロゲン受容体調節薬、たとえばタモキシフェン(tamoxifen
)、アロマターゼ阻害薬、たとえばアナストロゾール、またはエストロゲンダウンレギュレーターで処置されていない患者の処置に特に有益な可能性がある。
【0109】
化合物(I)およびその塩類、たとえばジフマレート塩は、タキサン類、たとえばパクリタキセル(paclitaxel)またはドセタキセル(docetaxel)とも組み合わせて使用でき
る。この組合わせは乳癌の処置に有用な可能性がある。たとえば、erbB2の過剰発現が低い乳癌の処置に際して。用語「erbB2の過剰発現が低い(low over-expression of erbB2)」は、Her2の蛍光インサイチュハイブリダイゼーション(fluorescent in-situ hybridization, FISH)が陰性である腫瘍を表わす。「erbB2の過剰発現
が低い」腫瘍である具体的な腫瘍は下記のものである:
(i)免疫組織学化学(immunohistochemistry, IHC)によりHer2 +;および/または
(ii)IHCによりHer2 ++、かつHer2蛍光インサイチュハイブリダイゼーション(FISH)陰性。
【実施例】
【0110】
本発明を以下の実施例によってさらに説明するが、それらは本発明の幾つかの態様を詳述するためのものである。これらの実施例は本発明の範囲を限定するためのものではなく、そのように解釈すべきでもない。ここに具体的に記載したもの以外の形で本発明を実施できることは明らかであろう。本明細書中の教示からみて本発明の多数の改変および変更が可能であり、それらは本発明の範囲に含まれる。
【0111】
実施例中において別途記載しない限り、下記のとおりである。
【0112】
(i)収率は、説明のために示したにすぎず、必ずしも達成可能な最大値ではない。
【0113】
(ii)融点は、Mettler DSC820e装置を用いてDSC分析により測定した。つまり、
1〜2mgの試料を精確に秤量し、ベント付き試料皿内で分析した(加熱を10℃/分で25℃から325℃まで行なった)。別途記載しない限り、本明細書中の融点はDSCを用いて測定した融解吸熱の開始温度を表わす。
【0114】
(iii)質量分析は70電子ボルトの電子エネルギーにより化学イオン化(CI)モードで直接曝露プローブを用いて行なった。指示した場合には、イオン化を電子衝突(EI)、高速原子衝撃(FAB)またはエレクトロスプレー(ESP)により行なった。m/zに関する数値を示す。一般に親質量を指示するイオンのみを報告する。別途記載しない限り、引用した質量イオンは(M+H)+であり、これはプロトン化質量イオンを表わす。M+という表示は電子の喪失により生じた質量イオンに対するものである。(M−H)−という表示はプロトンの喪失により生じた質量イオンに対するものである。
【0115】
(iv)NMRデータを示した場合、それは主診断プロトンについてのデルタ値の形であり、内標準物質としてのテトラメチルシラン(TMS)と対比した百万分率(ppm)で示され、別途指示しない限り、500MHzでペルジュウテリオジメチルスルホキシド(DMSO−d6)を溶媒として用いて測定した。以下の略号を用いた:s,一重線;d,二重線;t,三重線;q,四重線;m,多重線;br(broad),ブロード。
【0116】
(v)化学記号はそれらの通常の意味を有する。SI単位ないし記号を用いる。
【0117】
(vi)溶媒比は体積:体積(v/v)により示される。
【0118】
[実施例1] 4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリン(化合物(I))の製造
化合物(I)を以下に示すスキームに従って製造した。
【0119】
【化16】
【0120】
(工程1) 4−(5−シアノ−2−メトキシフェノキシ)ピペリジン−1−カルボン酸tert−ブチル(中間体2)の製造。
【0121】
3−ヒドロキシ−4−メトキシベンゾニトリル(化合物(X),6.00g,39.62mmole)、(4−メタンスルホニルオキシ)ピペリジン−1−カルボン酸tert−ブチル(16.6g,59.44mmoles)(Chemical & Pharmaceutical Bulletin 2001, 49(7), 822-829)、および炭酸カリウム(6.71g,47.55mmoles
)をイソプロパノール(78.98g)に懸濁し、混合物を撹拌しながら加熱還流した。
さらに(4−メタンスルホニルオキシ)ピペリジン−1−カルボン酸tert−ブチル(2.08g,7.43mmoles)を添加して反応を促進し、完了させた。次いで混合物を水(100.47g)の添加により冷却し、停止させた。中間体2の種晶を添加し、続いて0℃に冷却すると結晶質生成物が生成し、これを濾過により単離した。フィルターケークを水(8.86g)とイソプロパノール(6.97g)の混合物、続いて水(23.64g)で洗浄し、次いで乾燥させて、中間体2を得た(10.75g,80%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 1.48 (m, 2 H) 1.88 (m, 2 H) 3.13 (m, 2 H) 3.67 (m, 2 H) 3.83 (s, 3 H) 4.56 (tt, J=8.1, 3.8 Hz, 1 H) 7.13 (d, J=8.4 Hz, 1 H) 7.42 (dd, J=8.4, 1.9 Hz, 1 H) 7.51 (d, J=1.9 Hz, 1 H); 質量分析 m/z (M + H)+ 333.1。
【0122】
(工程2) 4−メトキシ−3−(ピペリジン−4−イルオキシ)ベンゾニトリル(化合物(VI))の製造。
【0123】
中間体2(39.31g,118.26mmoles)をエタノール(155.53g)に懸濁し、40℃に加熱した。このスラリーにHCl(46.61g,573.04mmoles)を徐々に添加した。混合物を60℃に加熱し、3時間保持した。反応混合物を20℃に冷却し、種晶を装入して結晶化を開始させた。生成した固体を0℃で濾過により単離し、エタノール(62.21g)で2回洗浄し、次いで乾燥させて化合物(VI)を塩酸塩として得た(29.84g,77%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.84 (m, 2 H) 2.09 (m, 2 H) 3.02 (ddd, J=12.7, 8.9, 3.4 Hz, 2 H) 3.20 (m, 2 H) 3.84 (s, 3 H) 4.63 (tt, J=7.7, 3.6 Hz, 1 H) 7.15 (d, J=8.5 Hz, 1 H) 7.45 (dd, J=8.5, 1.9 Hz, 1 H) 7.56 (d, J=1.9 Hz, 1 H) 9.16 (br. s, 2 H); 質量分析 m/z (M +
H)+ 233.2。
【0124】
(工程3) 2−[4−(5−シアノ−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(V))の製造。
【0125】
化合物(VI)(28.36g,95.82mmoles)、2−クロロ−N−メチルアセトアミド(12.37g,114.98mmoles)および炭酸カリウム(33.11g,239.55mmoles)を、アセトニトリル(161.36g)に懸濁した。反応混合物を3時間、加熱還流した。反応混合物を20℃に冷却し、水(386.26g)を装入した。反応物を75℃に加熱し、蒸留により体積を減らした。冷却すると結晶化が起きた。生成した固体を濾過により単離し、水で2回洗浄し(77.25gおよび128.75g)、次いで乾燥させて化合物(V)を得た(27.95g,94%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.68 (m, 2 H) 1.91 (m, 2 H) 2.29 (m, 2 H) 2.61 (d, J=4.7 Hz, 3 H) 2.67 (m, 2 H) 2.88 (s, 2 H) 3.83 (s, 3 H) 4.41 (tt, J=8.3,
4.0 Hz, 1 H) 7.11 (d, J=8.4 Hz, 1 H) 7.40 (dd, J=8.4, 1.9 Hz, 1 H) 7.47 (d, J=1.9 Hz, 1 H) 7.68 (q, J=4.7 Hz, 1 H); 質量分析 m/z (M + H)+ 304.2。
【0126】
(工程4) 2−[4−(5−シアノ−2−メトキシ−4−ニトロフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(IV))の製造。
【0127】
化合物(V)(8.78g,26.11mmoles)を酢酸(22.82g,364.87mmoles)に懸濁し、得られた反応混合物を5℃に冷却した。これに、温度を30℃より低く維持しながら硫酸(23.64g,234.95mmoles)を添加した。得られた溶液に硝酸(2.40g,26.63mmoles)を添加した。次いで反応混合物を35℃に加熱し、3時間保持した。さらに硝酸(117mg,1.31mmoles)および硫酸(1.31g,13.1mmoles)を装入し、反応混合物を35℃で30分間加熱した。この溶液を20℃に冷却し、アンモニア水(92.45g,1.
36moles)で停止させると、温度が50℃に上昇した。得られたスラリーに、プロピオニトリル(61.58g,1.12moles)および水(19g)を添加した。反応混合物を80℃に加熱すると透明な溶液が得られ、これは沈降すると2層になった。下層を分離した。反応混合物を20℃に冷却すると濃厚なスラリーが得られた。固体を濾過により単離し、プロピオニトリル(6.16g,112.0mmoles)で洗浄し、乾燥させて化合物(IV)を得た(7.44g,82%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.72 (m, 2 H) 1.97 (m, 2 H) 2.35 (m, 2 H) 2.61 (d, J=4.7 Hz, 3 H) 2.66 (m, 2 H) 2.90 (s, 2 H) 3.96 (s, 3 H) 4.73 (tt, J=8.4, 4.0 Hz, 1 H) 7.71 (q, J=4.7 Hz, 1 H) 7.82 (s, 1 H) 7.86 (s, 1 H). 質量分析 m/z (M + H)+ 349.2。
【0128】
(工程5) 2−[4−(4−アミノ−5−シアノ−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(III))の製造。
【0129】
化合物(IV)(7.42g,19.38mmoles)を水(44.52g)およびメタノール(5.35g)に懸濁した。これに亜ジオチン酸ナトリウム(11.91g,58.15mmoles)を添加し、得られた反応混合物を60℃に加熱した。反応混合物に塩酸(46.98g,463.89mmoles))を添加すると溶液が得られ、これを60℃に3時間保持した。次いで反応混合物を20℃にまで放冷した。水酸化ナトリウム水溶液(15.51g,182.2mmoles)、続いて2−メチルテトラヒドロフラン(58.0g)を装入した。反応混合物を60℃に加熱し、これは沈降すると2層になり、下層を廃棄した。反応混合物の体積を真空蒸留により減らし、メチル tert−ブチルエーテル(18.54g)を添加するとスラリーが得られ、これを10℃に冷却し、次いで固体を濾過により採集した。固体を2−メチルテトラヒドロフラン(5.8g)で洗浄し、乾燥させて化合物(III)を得た(5.4g,78%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.62 (m, 2 H) 1.82 (m, 2 H) 2.20 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.65 (m, 2 H) 2.86 (s, 2 H) 3.72 (s, 3 H) 4.00 (tt, J=8.3, 4.0 Hz, 1 H) 5.66 (br. s, 2 H) 6.39 (s, 1 H) 6.94 (s, 1 H) 7.65 (q, J=4.7 Hz, 1 H). 質量分析 m/z (M + H)+ 319.2。
【0130】
(工程6) 2−[4−(5−シアノ−4−{[(ジメチルアミノ)メチレン]アミノ}−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(II))の製造。
【0131】
化合物(III)(18.21g,52.05mmoles)を2−メチルテトラヒドロフラン(99.62g)に懸濁した。これに酢酸(162.79mg)およびN,N−ジメチルホルムアミドジメチルアセタール(DMA)(8.63g,70.27mmoles)を添加し、得られた反応混合物を76℃で16時間加熱した。さらにN,N−ジメチルホルムアミドジメチルアセタール(639.41mg,5.20mmoles)を反応混合物に添加して、反応を確実に完了させた。反応混合物を30℃に冷却すると、その間に結晶化が起きた。生成した固体を濾過により単離し、2−メチルテトラヒドロフラン(14.23g)で洗浄して、化合物(II)を得た(19.53g,97%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.65 (m, 2 H) 1.86 (m, 2 H) 2.24 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.66 (m, 2 H) 2.87 (s, 2 H) 2.95 (s, 3 H) 3.04 (s, 3 H) 3.81 (s, 3 H) 4.19 (tt, J=8.2, 3.8 Hz, 1 H) 6.72 (s, 1 H) 7.15 (s, 1 H) 7.67 (q, J=4.7 Hz, 1 H) 7.90 (s, 1 H); 質量分析 m/z (M + H)+ 374.2。
【0132】
(工程7) 化合物(I)の製造。
【0133】
2−[4−(5−シアノ−4−{[(ジメチルアミノ)メチレン]アミノ}−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(II),
7.00g,17.71mmoles)をメトキシベンゼン(35.8g)に懸濁した。酢酸(16.6g)を装入し、得られた溶液に3−クロロ−2−フルオロアニリン(2.71g,18.07mmoles)を添加した。反応混合物を90℃で20時間加熱し、次いで20℃に冷却した。水(37.04g)を反応混合物に装入し、有機層を廃棄した。得られた水性混合物にイソプロパノール(39.00g)、続いてアンモニア水(20.79g,25%)を装入した。反応混合物を30℃に加熱し、化合物(I)の種晶を添加すると、これにより結晶化が誘発された。次いで反応物を0℃に冷却し、生成物を濾過により単離した。フィルターケークを水(7.28g)とイソプロパノール(4.68g)の混合物で2回洗浄し、次いで乾燥させて化合物(I)を得た(5.65g,55%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (m, 2 H) 2.04 (m, 2 H) 2.38 (m, 2 H) 2.62 (d, J=4.5 Hz, 3 H) 2.74 (m, 2 H) 2.94 (s, 2 H) 3.93 (s, 3 H) 4.56 (tt, J=8.1, 3.8 Hz, 1 H) 7.21 (s, 1 H) 7.28 (m, 1 H) 7.50 (m, 2 H) 7.73 (q, J=4.5 Hz, 1 H) 7.81 (s, 1 H) 8.36 (s, 1 H) 9.56 (br.s, 1 H); 質量分析 m/z (M + H)+ 474.2, 476.2。
【0134】
[実施例2] 4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリン(化合物(I))の製造
化合物(I)を以下に示すスキームに従って製造した。
【0135】
【化17】
【0136】
工程1、2、3および4は実施例1に示したとおりである。
【0137】
(工程5,別法1) 化合物(III)の製造。
【0138】
2−[4−(5−シアノ−2−メトキシ−4−ニトロフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(IV),15.00g,42.50mmoles)を水(90.00g)およびメタノール(59.38g)に懸濁した。これに亜ジオチン酸ナトリウム(30.47g,148.75mmoles)および水(90.00g)を添加し、得られた反応混合物を30℃に加熱し、2時間保持した。反応混合物に塩酸(27.98g,276.25mmoles))を添加すると溶液が得られ、これを60℃に2時間保持した。水酸化ナトリウム水溶液(30.60g,382.49mmoles)、続いてライン洗浄(line wash)水(30.00g)を添加した。反応混合物を2
5℃に冷却するとスラリーが得られ、これを濾過により採集した。固体を水(30.00g)で洗浄し、乾燥させて化合物(III)を得た(13.50g,82%の収率); 1H
NMR (400 MHz, DMSO-d6) δ ppm 1.62 (m, 2 H) 1.82 (m, 2 H) 2.20 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.65 (m, 2 H) 2.86 (s, 2 H) 3.72 (s, 3 H) 4.00 (tt, J=8.3, 4.0 Hz, 1 H) 5.66 (br. s, 2 H) 6.39 (s, 1 H) 6.94 (s, 1 H) 7.65 (q, J=4.7 Hz, 1 H). 質量分析 m/z (M+H)+ 319.2。
【0139】
(工程5,別法2) 化合物(III)の製造。
【0140】
化合物(IV)(8.00g,22.67mmoles)およびカーボン上1%白金+2%バナジウム触媒(1.23g,0.023mmoles)をアセトニトリル(94.00g)に懸濁した。反応混合物を3Bar Gの圧力で35℃の温度において3時間水素化した。完了した時点で反応混合物を濾過して触媒を分離し、アセトニトリル(31.33g)で洗浄した。反応混合物の体積を真空蒸留により減らすとスラリーが得られ、これを0℃に冷却し、次いで固体を濾過により採集した。固体をアセトニトリル(12.53g)で洗浄し、乾燥させて化合物(III)を得た(5.88g,78%の収率); 1H
NMR (400 MHz, DMSO-d6) δ ppm 1.62 (m, 2 H) 1.82 (m, 2 H) 2.20 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.65 (m, 2 H) 2.86 (s, 2 H) 3.72 (s, 3 H) 4.00 (tt, J=8.3, 4.0 Hz, 1 H) 5.66 (br. s, 2 H) 6.39 (s, 1 H) 6.94 (s, 1 H) 7.65 (q, J=4.7 Hz, 1 H). 質量分析 m/z (M+H)+ 319.2。
【0141】
(工程6) N,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミド(化合物(XI))の製造。
【0142】
3−クロロ−2−フルロアニリン(51.21g,341.22mmoles)をシクロヘキサン(87.07g)に懸濁した。これにオルトギ酸エチル(22.28g,150.32mmoles)および酢酸(0.94g,15.03mmoles)を添加した。得られた反応混合物を撹拌しながら48℃に12時間加熱した。これに続いて反応混合物を12時間かけて20℃に冷却し、固体生成物を濾過により単離した。フィルターケークをシクロヘキサン(26.12g)で洗浄し、40℃で真空乾燥して、化合物(XI)を白色結晶質生成物として得た(33.95g,93%の収率); 1H NMRスペクトル (400 MHz, DMSO-d6) δ ppm 7.14 (t, 2 H) 7.22 (m, 2 H) 8.14 (s, 1 H), 9,98 (s, 1 H);
質量分析(by GC-MS EI): m/z (M+) 300.0。
【0143】
(工程7,別法1) 化合物(I)の製造。
【0144】
2−[4−(4−アミノ−5−シアノ−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(III))(10g,29.84mmol)およびN,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミド(化合物(XI))(11.46g,37.3mmol)を2−メチルテトラヒドロフラン(30.4ml)に懸濁し、80℃に加熱した。この黄色懸濁液に酢酸(7.6ml,127.33mmol)を添加し、得られた溶液を92℃に6時間加熱した。2−メチルテトラヒ
ドロフラン(66.5ml)および水(28.5ml)を添加し、混合物を55℃に冷却した後、50% w/w水酸化ナトリウム(7ml,131.29mmol)を添加すると温度が63℃に上昇した。温度をさらに69℃に高め、沈降後に水層を廃棄した。有機層を水で洗浄し(3×20ml)、それぞれ水層を沈降後に廃棄した。2−メチルテトラヒドロフラン(100ml,997mmol)を添加し、蒸留により体積を減らした。種晶を添加して結晶化を誘発し、得られた混合物を15℃に冷却した。上記実験から自然結晶化に伴って結晶質形態が最初に得られた。生成した固体を濾過により単離し、2−メチルテトラヒドロフラン(19ml)で2回洗浄し、40℃で真空乾燥して、化合物(I)を白色固体として得た(12.14g,95%); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.12 (d, J= 6Hz,1.3H), 1.26 -1.36 (m, 0.4H), 1.75-1.97 (m, 3.3H), 2.02-2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.7Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H),
3.52-3.59 (m, 0.4H), 3.72-3.87 (m, 0.86H), 3.95 (s, 3H), 4.53-4.63 (m, 1H), 7.22 (s, 1H), 7.29 (dt J= 1Hz J= 8Hz, 1H), 7.51 (dt J= 7.4Hz, J= 18Hz, 2H), 7.71-7.77 (m, 1H), 7.82 (s, 1H), 8.37 (s, 1H), 9.57 (s, 1H). 質量分析 m/z (M+H)+ 474.0
。上記のNMRデータは0.43モル当量で存在する2−メチルテトラヒドロフラン溶媒に関する信号をも含む。溶媒に関係する信号はδ ppmシフト1.12, 1.26-1.36, 3.52-3.59および3.72-3.87にあるものである。1.75-1.93にあるクラスターは溶媒と親化合物に関する信号を含む。この化合物のXRPDを図2に示す。
【0145】
(工程7,別法2) 化合物(I)の製造。
【0146】
化合物(III)(15g,44.76mmol)および化合物(XI)(17.19g,55.95mmol)を2−メチルテトラヒドロフラン(45.6ml)に懸濁し、83℃に加熱した。この黄色懸濁液に酢酸(11.4ml,190.99mmol)を添加し、得られた溶液を92℃に3.5時間加熱した。2−メチルテトラヒドロフラン(105ml)および水(50ml)を添加し、混合物を49℃に冷却した後、50% w/w水酸化ナトリウム(10.74ml,201.4mmol)を添加すると温度が62℃に上昇した。温度を62℃に保持し、沈降後に水層を廃棄した。有機層を水で洗浄し(3×30ml)、沈降後にそれぞれ水層を廃棄した。混合物を15℃に冷却し、種晶を添加して結晶化を誘発した。上記実験から自然結晶化に伴って結晶質形態が最初に得られた。生成した固体を濾過により単離し、2−メチルテトラヒドロフラン(21ml)で2回洗浄し、40℃で真空乾燥して、化合物(I)を白色固体として得た(20.12g,95%). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.75-1.86 (m, 2H), 2.02-2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.7Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.95 (s, 3H), 4.53-4.63 (m, 1H), 7.22 (s, 1H), 7.29 (dt J= 1Hz J= 8Hz, 1H), 7.51 (dt J=
7.4Hz, J= 18Hz, 2H), 7.71-7.77 (m, 1H), 7.82 (s, 1H), 8.37 (s, 1H), 9.57 (s, 1H). 質量分析 m/z (M+H)+ 474.0。この化合物のXRPDを図3に示す。
【0147】
(工程7,別法3) 化合物(I)の製造。
【0148】
化合物(III)(15.1g,45.06mmol)および化合物(XI)(17.31g,56.32mmol)を2−メチルテトラヒドロフラン(46ml)に懸濁し、80℃に加熱した。この黄色懸濁液に酢酸(12ml,458mmol)を添加し、得られた溶液を92℃に7時間加熱した。2−メチルテトラヒドロフラン(100ml)および水(43ml)を添加し、混合物を59℃に冷却した後、50% w/w水酸化ナトリウム(11ml,207mmol)を添加すると温度が71.5℃に上昇した。温度を69℃に調整し、沈降後に水層を廃棄した。有機層を水で洗浄し(2×43ml)、沈降後にそれぞれ水層を廃棄した。2−メチルテトラヒドロフラン(72ml)を大気圧蒸留により除去し、イソプロピルアルコール(72ml)の添加により交換した。さらに72mlの溶媒を大気圧蒸留により除去し、イソプロピルアルコール(72ml)により交換し
た。種晶を添加して結晶化を誘発し、得られた混合物を15℃に冷却した。固体を濾過により単離し、イソプロピルアルコール(32ml)で2回洗浄し、40℃で真空乾燥して、化合物(I)を白色固体として得た(20.86g,87%). 1H NMR (400 MHz, DMSO-d6)δ ppm 1.04 (d, J= 6Hz, 6H),1.75-1.88 (m, 2H), 2.02-2.15 (m, 2H), 2.35-2.44
(m, 2H), 2.64 (d, J= 4.7Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.73-3.84 (m,
1H), 3.95 (s, 3H), 4.34 (d, J = 4.2Hz, 1H), 4.53-4.63 (m, 1H), 7.22 (s, 1H), 7.29 (dt J= 1Hz J= 8Hz, 1H), 7.51 (dt J= 7Hz, J= 18Hz, 2H), 7.71-7.77 (m, 1H), 7.82 (s, 1H), 8.37 (s, 1H), 9.57 (s, 1H). 質量分析 m/z (M+H)+ 474.0。NMRデータは存在する1モル当量のイソプロパノールに関する信号を含む。この化合物のXRPDを図4に示す。
【0149】
[実施例3] 4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリン ジ−[(2E)−ブタ−2−エンジオエート](化合物(I)ジフマレート塩)の製造
化合物(I)ジフマレート塩を以下に示すスキームに従って製造した。
【0150】
【化18】
【0151】
工程1、2、3、4、5および6は実施例2に示したとおり実施した。
【0152】
(工程7) 化合物(I)ジフマレート塩の製造。
【0153】
化合物(III)(17.90mmoles)およびN,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミド(化合物(XI))(7.04g,23.27mmoles)をtert−ブチルアルコール(88.95g)に懸濁した。この懸濁液にフマル酸(10.39g,89.52mmoles)を添加し、混合物を撹拌しながら80℃に2.5時間加熱した。水(11.40g,632.80mmoles)を装入し、反応をさらに21.5時間続けた。反応物を12時間かけて20℃にまで冷却すると、その間に結晶化が起きた。生成した固体を濾過により単離し、水(1.00)とtert−ブチルアルコール(7.80g)の混合物で洗浄し、続いて水(0.50g)とtert−ブチルアルコール(7.30g)の混合物で洗浄した。固体を40℃で真空乾燥して、化合物(I)ジフマレート塩(8.17g,61.40%)をマスタード黄色粉末として得た; 1H NMR (400 MHz, DMSO-d6) δ ppm 1.83 (m, 2 H, broad) 2.07 (m, 2 H, broad) 2.64 (d, J=5.0 Hz, 3 H) 2.80 (m, 2 H, broad) 3.03 (s, 2 H) 3.94 (s, 3 H) 4.58
(m, 1 H) 6.63 (s, 4 H) 7.22 (s, 1 H) 7.29 (td, J=8.5, 1.0 Hz, 1 H) 7.51 (m, 2 H) 7.82 (m, 2 H) 8.37 (s, 1 H); 質量分析 m/z (M+H)+ 474.0。
【0154】
[実施例4] 4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリン(化合物(I))の製造
化合物(I)を以下に示すスキームに従って製造した。
【0155】
【化19】
【0156】
工程1、2、3、4および5は実施例2に示したとおり実施した。
【0157】
(工程6) N’−(3−クロロ−2−フルオロ−フェニル)−N,N−ジメチル−ホルムアミジン(化合物(XII))の製造。
【0158】
3−クロロ−2−フルロアニリン(5.30g,35.29mmoles)を2−メチルテトラヒドロフラン(52.94g)に溶解した。これにN,N−ジメチルホルムアミドジメチルアセタール(6.07g,49.41mmoles)および酢酸(0.11g,1.76mmoles)を添加した。得られた反応混合物を撹拌しながら76℃に3時間加熱した。これに続いて溶液を40℃で真空下に除去して、化合物(XII)を黄色の油として得た(6.60g,93%の収率); 1H NMRスペクトル (400 MHz, DMSO-d6) δ
ppm 2.74 (s, 0.29H), 2.89 (s, 0.31H), 2.94 (s, 2.75H), 3.03 (s, 2.66H), 3.34 (br s, 0.70H), 5.48 (s, 0.06H) 6.91-7.10 (m, 3H), 7.79 (s, 1 H), 7.96 (s, 1 H)。上記のNMRデータは0.06モル当量で存在するN,N−ジメチルホルムアミドジメチルアセタールに関する信号を含む。N,N−ジメチルホルムアミドジメチルアセタールに関係する信号はδ ppmシフト3.75、および6.90-6.95にあるものである。δ ppm 3.35にある信号は残留水によるものである。質量分析(LCMS EIによる): m/z (M+H)+ 201.2。
【0159】
(工程7) 化合物(I)の製造。
【0160】
2−[4−(4−アミノ−5−シアノ−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(III))(0.50g,1.45mmol)およびN’−(3−クロロ−2−フルオロ−フェニル)−N,N−ジメチル−ホルムアミジン(化合物(XII))(0.32g,1.52mmol)を、メトキシベンゼン(3.1ml)に懸濁した。この黄色懸濁液に酢酸(1.52ml,25.51mmol)を添加し、得られた溶液を90℃に14時間加熱した。反応混合物を20℃に冷却し、水(2.58mL)を添加した。有機層を除去し、水層をメトキシベンゼン(1.4mL)で洗浄した。エタノール(2.45mL)およびアンモニア(1.94ml,25.55mmoles)を水層に添加した。この溶液を90℃に加熱すると、若干のエタノールが蒸発により失われた。この溶液を40℃に冷却した。種晶を添加して結晶化を誘発し、得られた混合物を20℃に冷却した。固体を濾過により単離して、化合物(I)を白色固体として得た(0.61g,73%の収率). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.75-1.87
(m, 2H), 2.02-2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.8Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.35 (s, 5.4H), 3.75 (s, 1.3H), 3.95 (s, 3H), 4.58 (hept., J=4.0Hz, 1H), 6.90-6.95 (m, 1.3H), 7.23 (s, 1.8H), 7.26-7.34 (m, 1H), 7.45-7.58 (m 2H), 7.72-7.78 (m, 1H), 7.83 (s, 1H), 8.38 (s, 1H), 9.58 (s, 1H)。上記の
NMRデータは0.40モル当量で存在するメトキシベンゼン溶媒に関する信号を含む。この溶媒に関係する信号はδ ppmシフト3.75、および6.90-6.95にあるものである。7.26-7.34にあるクラスターは溶媒と親化合物に関する信号を含む。δ ppm 3.35にある信号は
残留水によるものである。質量分析 m/z (M + H)+ 474.0, 476.0。
【0161】
[実施例5] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエート]形態Aの製造
フマル酸(2.7g,23.22mmol)のメタノール(95ml)中における溶液を、2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(I))(5.62g,89% w/wのもの,10.55mmol)のイソプロパノール(100ml)中における混合物に、温度を>65℃に維持しながら添加した。混合物を澄明になるまで1時間、加熱還流した。反応混合物を90分間かけて30℃にまで冷却し、30分間保持して結晶化を確立した。反応物を2時間かけて0℃にまで冷却し、1時間保持した後、濾過により単離した。フィルターケークを冷イソプロパノールで2
回洗浄し(2×10ml)、50℃で真空乾燥して、表題化合物を白色固体として得た(5.84g,78%); 1H NMRスペクトル: (DMSO) 1.85 (m, 1H), 2.08 (m, 1H), 2.50 (m, 1H), 2.66 (d, 3H), 2.83 (m, 1H), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, 1H), 6.64 (s, 4H), 7.23 (s, 1H), 7.28 (m, 1H), 7.46 (ddd, 1H), 7.55 (m, 1H), 7.70 (broad q, 1H), 7.85 (s, 1H), 8.38 (s, 1H)。
【0162】
[実施例6] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエート]形態Aの製造
フマル酸(1.4kg,12.1mol)のメタノール(26.6kg)中における溶液を、2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(2.93kg,84.8% w/w,5.24mol)のイソプロパノール(39kg)中における混合物に、温度を>65℃に維持しながら添加した。ライン洗浄メタノール(3.6kg)を装入した。混合物を澄明になるまで1時間、加熱還流し、続いてライン洗浄メタノール(7kg)を添加した。反応混合物を大気圧蒸留して47kgの留出物を除去した。イソプロパノール(15.8kgを添加し、反応混合物を蒸留して15.6kgの留出物を除去した。蒸留中に結晶化が起きた。イソプロパノール(21kg)を添加し、反応物を8時間かけて0℃に冷却し、1時間保持した後、濾過により単離した。フィルターケークを、冷却した50:50イソプロパノール:MeOH(4kg)、続いて冷イソプロパノール(4kg)で洗浄し、50℃で真空乾燥して、表題化合物を白色固体として得た(3.64kg,98%); 1H NMRスペクトル: (DMSO) 1.85 (m, 1H), 2.08 (m, 1H), 2.50 (m, 1H), 2.66 (d, 3H), 2.83 (m, 1H), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, 1H), 6.64 (s, 4H), 7.23 (s, 1H), 7.28 (m, 1H), 7.46 (ddd, 1H), 7.55 (m, 1H), 7.70 (broad q, 1H), 7.85 (s, 1H), 8.38 (s, 1H)。
【0163】
[実施例7] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエート]形態Aの製造
2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(I))(60.19g,88% w/wのもの,111.8mmol)を酢酸エチル(1550ml)に溶解した。この溶液を濾過により澄明化し、フィルターを酢酸エチル(53ml)で洗浄した。この溶液を40℃に冷却した。次いで、フマル酸(26.60g,257.0mmol)のイソプロパノール(408ml)中における澄明化した溶液を1時間かけて添加した。フマル酸溶液を澄明化するために用いたフィルターを、次いでイソプロパノール(37ml)で洗浄した。40℃に1時間保持した後、反応物を1時間かけて20℃にまで冷却した。反応混合物を13.5時間保持した後、生成物を濾過により単離した。フィルターケークを酢酸エチル(82ml):イソプロパノール(24ml)で2回洗浄し、次いで40℃で真空乾燥して、表題化合物を白色固体として得た(72.32g,90%); 1H NMRスペクトル: (DMSO) 1.85 (m, 1H), 2.08 (m, 1H), 2.50 (m, 1H), 2.66 (d, 3H), 2.83 (m, 1H), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, 1H), 6.64 (s, 4H), 7.23 (s, 1H), 7.28 (m, 1H), 7.46 (ddd, 1H), 7.55 (m, 1H), 7.70 (broad q, 1H), 7.85 (s, 1H), 8.38 (s, 1H)。
【0164】
[実施例8] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエ
ート]形態Aの製造
2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(I))(2.75g,推定100% w/wのもの,5.80mmol)を、酢酸エチル(94ml)およびイソプロパノール(14ml)に溶解した。溶液を蒸留し、これにより25.2mlの留出物を採集した。溶液を40℃に冷却した。フマル酸(1.38g,11.90mmol)のイソプロパノール(21ml)中における澄明化した溶液を、次いで1時間かけて添加した。化合物(I)ジフマレート形態Aの種晶(3.7mg,5.3μmol)を添加した。フマル酸溶液を澄明化するために用いたフィルターを、次いでイソプロパノール(2ml)で洗浄した。40℃に1時間保持した後、反応物を2時間かけて20℃にまで冷却した。反応混合物を15時間保持した後、生成物を濾過により単離した。フィルターケークを酢酸エチル(4.3ml):イソプロパノール(1.2ml)で2回洗浄し、次いで40℃で真空乾燥して、表題化合物を白色固体として得た(72.32g,90%); 1H NMRスペクトル: (DMSO) 1.85 (m, 1H), 2.08 (m, 1H), 2.50 (m, 1H), 2.66 (d, 3H), 2.83 (m, 1H), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, 1H),
6.64 (s, 4H), 7.23 (s, 1H), 7.28 (m, 1H), 7.46 (ddd, 1H), 7.55 (m, 1H), 7.70 (broad q, 1H), 7.85 (s, 1H), 8.38 (s, 1H)。
【0165】
[実施例9] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエート]形態Aの製造
2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(I))(1g,1.86mmoles)およびフマル酸(0.44g,3.81mmoles)を水(4.4g)に懸濁し、85℃に加熱した。反応混合物を1℃/分で60℃にまで冷却し、温度が77℃になった時点で化合物(I)形態Aの種晶を添加した。生成した固体を濾過により単離し、アセトンで2回洗浄し(1回の洗浄当たり0.70g)、40℃で真空乾燥して、表題化合物を得た(0.89g,68%の収率); 1H NMR (400 MHz, DMSO-d6) d ppm 1.84 (m, 2 H) 2.08 (m, 2 H) 2.55 (m, 2 H) 2.63 (d, J=4.7
Hz, 3 H) 2.86 (m, 2 H) 3.12 (s, 2 H) 3.93 (s, 3 H) 4.59 (tt, J=7.8, 3.7 Hz, 1 H) 6.62 (s, 4 H) 7.21 (s, 1 H) 7.27 (td, J=8.1, 1.3 Hz, 1 H) 7.49 (m, 2 H) 7.86 (m, 2 H) 8.36 (s, 1 H) 9.63 (br. s., 1 H)。
【0166】
化合物(I)ジフマレート形態Aはさらさらした粉末である。化合物(I)ジフマレートのX線粉末回折(図1)は、この物質が結晶質であることを示す。X線粉末回折分析は、シンチレーション検出器を備えたSiemens D5000粉末X線回折計を用いて実施した;X
線源は、波長1.54Åを生じるCu Kαであった;データを2−θ範囲2〜40°にわたって2−θ 0.02°の増分で、増分当たり1秒を用いて収集し、次表に同定するカテゴリーに分類した:
【0167】
【表1】
【0168】
X線粉末回折の技術分野の専門家には、ピークの相対強度が、たとえば試料の分析に影響を及ぼす30ミクロンを超えるサイズの結晶粒および不均一なアスペクト比により影響される可能性があることが分かるであろう。反射の位置は試料が回折計内で置かれた厳密な高さおよび回折計のゼロ目盛定めにより影響される可能性があることも当業者には分かるであろう。試料の表面平面性もわずかな影響をもつ可能性がある。したがって、提示した回折パターンデータは絶対値と解釈すべきではない(詳細な情報については、Jenkins, R & Snyder, R.L. ‘Introduction to X-Ray Powder Diffractometry’ John Wiley & Sons, 1996を参照)。
【0169】
[実施例10] 化合物(I)ジフマレートの錠剤配合物
粉末成分をミキサーに装入し、混合して薬物の均一な分散物を得る。結合剤溶液を調製し、粉末に添加し、適切な湿潤素材が形成されるまでさらに混合する。湿潤素材をスクリーンに通し、得られた顆粒を乾燥させて適宜な含水率にする。乾燥顆粒を適宜なサイズのスクリーンに通し、そしてステアリン酸マグネシウムとブレンドした後、一般的な錠剤製造装置により圧縮して錠剤コアにする。圧縮したコアを、一般的な有孔ドラムコーターによりフィルムコーティング成分の水性懸濁液でコートする。
【0170】
上記に従って製造した、2.5、10、40および100mgの化合物(I)に相当する化合物(I)ジフマレート形態Aを含有するフィルムコートした錠剤を、下記の表1に示す。
【0171】
【表2】
【0172】
錠剤を製造するのに適した製造方法を下記に概説する。
【0173】
【表3】
【0174】
本明細書に記載する化合物は遊離の非塩形で生じる可能性があり、あるいは塩類として生じる可能性があり、用語「化合物」の使用が遊離形の化合物および化合物の塩類を包含することは当業者には認識されるであろう。
【0175】
当業者に理解されるように、いずれかまたはすべての目的について、特に記述提示に関して、本明細書に開示するすべての範囲は、そのすべての可能な下位範囲および下位範囲の組合わせ、ならびにその範囲を構成する個々の数値、特に整数値をも包含する。列記した範囲はいずれも、その範囲が少なくとも2、3、4、5、6等分などされることを十分に記述および可能にすることは、容易に認識できる。限定ではない例として、本明細書中に述べる各範囲は、下1/3、中1/3、上1/3などに容易に分割できる。同様に当業者に理解されるように、「最高で(up to)」、「少なくとも(at least)」、「より大
きい(greater than)」、「より少ない(less than)」、「より多い(more than)」、「またはそれより多い(or more)」などの語はすべて、引用した数値を含み、さらに前
記に述べたように下位範囲に分割できる範囲を表わす。同様に、本明細書に開示するすべての比率は、その広い比率内に含まれるすべての下位比率をも含む。
【0176】
たとえばMarkushグループのようにメンバーを一般的な方法でグループにまとめた場合
、本発明はまとめて挙げたグループ全体だけでなくそのグループの各メンバー個々および
その主グループの可能なすべての下位グループをも包含することも、当業者には容易に認識されるであろう。さらに、すべての目的に関して、本発明は主グループだけでなく1以上のグループメンバーが存在しない主グループをも包含する。本発明は本発明内のいずれか1以上のグループメンバーの明らかな除外をも考慮する。
【0177】
当業者に理解されるように、成分量、特性、たとえば分子量、反応条件などを表わすものを含めたすべての数値は概数であり、すべての例において用語「約」で修飾されていると理解される。これらの数値は、本発明の教示を利用して当業者が求める目的特性に応じて変動する可能性がある。そのような数値は本来、それらのそれぞれの試験測定値にみられる標準偏差から必然的に生じる変動性を含むことも理解される。
【0178】
本明細書に開示するすべての参考文献を、本明細書に具体的に援用する。
【0179】
本明細書中の「工程」という表記、または「工程」に付けた番号は便宜的に用いたものにすぎず、本明細書中に述べる本発明を分類、規定または限定しない。
【0180】
特定の態様を説明および記載したが、これらの態様は本発明の範囲を限定しないこと、ならびに特許請求の範囲に定めた本発明の広い観点から逸脱することなく当業者が変更および改変しうることを理解すべきである。
【技術分野】
【0001】
本発明は、4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバメトイルミル)ピペリジン−4−イル]オキシ}キナゾリンまたはその塩(以下、「化合物(I)」)の製造方法、および化合物(I)の製造に用いる中間体に関する。
【背景技術】
【0002】
式(I)の化合物は、国際特許出願公開番号WO2005/028469に例1として開示され、以
下の構造である。
【0003】
【化1】
【0004】
化合物(I)はerbB受容体型チロシンキナーゼの阻害薬であり、特に化合物(I)はEGFRおよびerbB2受容体型チロシンキナーゼの有効な阻害薬である。化合物(I)は、リガンド刺激されたEGFR/erbB3および/またはerbB2/erbB3ヘテロ二量体化に続くerbB3リン酸化の阻害により、erbB3仲介シグナル伝達をも阻害する。化合物(I)は、癌などの過増殖性障害の処置に有用であると予想される。
【0005】
WO 03/082831には、種々の4−(3−クロロ−2−フルオロアニリノ)キナゾリン類の製造が開示されている。しかし、化合物(I)はそこに開示されていない。
【0006】
WO2005/028469には、例1として化合物(I)の製造法が以下のように開示されている
。
【0007】
2−クロロ−N−メチルアセトアミド(32mg,0.3mmol)を、4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−[(ピペリジン−4−イル)オキシ]キナゾリン(120mg,0.3mmol)、ヨウ化カリウム(16mg,0.1mmol)および炭酸カリウム(50mg,0.36mmol)のアセトニトリル(5ml)中における混合物に添加した。この混合物を1時間、加熱還流した。溶媒を真空下で蒸発させた後、残留物をジクロロメタンに装入した。この有機溶液を水およびブラインで洗浄し、硫酸マグネシウムで乾燥させた。溶媒を真空下で蒸発させた後、残留物をシリカゲル上でのクロマトグラフィー(溶離剤:ジクロロメタン中、1%−2%の7Nメタノール性アンモニア)により精製して、化合物(I)が得られた。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】WO 2005/028469
【特許文献2】WO 03/082831
【発明の概要】
【0009】
本発明者らは化合物(I)を製造するための別法を見出した。この方法は、少ない数の製造工程で高収率において不純物が最少である化合物(I)を提供する。したがってこの方法は、化合物(I)の大規模製造に使用するのに適している。
【0010】
1つの態様は、4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリンまたはその塩の製造方法であって:
(a)式(II)の化合物:
【0011】
【化2】
【0012】
を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させること;または
(b)式(III)の化合物:
【0013】
【化3】
【0014】
を適切な酸の存在下で式(XI)または式(XII)の化合物:
【0015】
【化4】
【0016】
と反応させること;
を含む方法を提供する。
【図面の簡単な説明】
【0017】
【図1】図1は、化合物(I)ジフマレート形態AについてのX線粉末回折パターン(XRPD)を示す。x軸は2θ値を示し、y軸はカウントを示す。
【図2】図2は、2−メチルテトラヒドロフラン溶媒和物としての化合物(I)のX線粉末回折パターン(XRPD)を示す。x軸は2θ値を示し、y軸はカウントを示す。
【図3】図3は、水和物としての化合物(I)のX線粉末回折パターン(XRPD)を示す。x軸は2θ値を示し、y軸はカウントを示す。
【図4】図4は、イソプロパノール溶媒和物としての化合物(I)のX線粉末回折パターン(XRPD)を示す。x軸は2θ値を示し、y軸はカウントを示す。
【発明を実施するための形態】
【0018】
1態様は、4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリンまたはその塩の製造方法であって:
(a)式(II)の化合物:
【0019】
【化5】
【0020】
を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させることを含む方法を提供する。
【0021】
この反応は、適切には、適切な酸、たとえば酢酸、ブタン二酸、プロパン酸、コハク酸、フマル酸およびクエン酸、またはその混合物から選択される1種類以上の酸の存在下で実施される。本発明の1態様において、酸は酢酸である。
【0022】
この反応は、適切には、不活性溶媒、たとえばシクロヘキサン、芳香族炭化水素系溶媒、たとえばトルエン、メトキシベンゼンもしくはキシレン;ニトリル系溶媒、たとえばアセトニトリル;エーテル類、たとえば2−メチルテトラヒドロフラン;またはエステル、たとえば酢酸イソプロピルの存在下で実施される。1態様において、溶媒はトルエン、シクロヘキサン、メトキシベンゼン、キシレン、アセトニトリル、2−メチルテトラヒドロフランおよび酢酸イソプロピルから選択される。他の態様において、溶媒はトルエン、シクロヘキサン、メトキシベンゼンおよびキシレンから選択される。さらに他の態様において、溶媒はメトキシベンゼンである。
【0023】
この反応は、適切には高められた温度、たとえば約80から約120℃まで、たとえば約90〜120℃、適切には約90℃で実施される。
【0024】
適切には、式(II)の化合物に対して等モルまたは過剰モルの3−クロロ−2−フルオロアニリンを使用する。たとえば、3−クロロ−2−フルオロアニリンと式(II)の化合物のモル比は、約1:1から約1:2まで、適切には約1:1である。
【0025】
いずれの態様においても、化合物(I)を常法により単離することができる。たとえば、実施例に記載するように化合物(I)を水中に抽出し、そして溶液から結晶化させることができる。必要であれば、溶液からの化合物(I)の結晶化はその溶液に化合物(I)の種晶を添加することにより開始できる。生成した固体を次いで常法により、たとえば化合物(I)の濾過および乾燥により採集することができる。
【0026】
本発明のさらに他の観点によれば、式(II)の化合物の製造方法であって、式(III)の化合物:
【0027】
【化6】
【0028】
をN,N−ジメチルホルムアミドジメチルアセタールと反応させることを含む方法が提供される。
【0029】
この反応は、適切には酸性条件下で実施される。たとえば、この反応は適切には酢酸の存在下で実施される。
【0030】
この反応は、好都合には適切な溶媒、たとえばエーテル類、たとえば2−メチルテトラヒドロフラン、または芳香族炭化水素、たとえばトルエンの存在下で実施される。この反応は、適切には高められた温度、たとえば約70〜105℃、適切には約76℃で実施される。
【0031】
他の態様において、化合物(I)は、式(III)の化合物をN,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミド(化合物(XI))と反応させることにより、式(III)の化合物から直接製造できる。この反応は、適切には酸性条件下で実施される。この反応は、適切には、適切な酸、たとえば酢酸、ブタン二酸、プロパン酸、コハク酸、フマル酸およびクエン酸、またはその混合物から選択される1種類以上の酸の存在下で実施される。本発明の1態様において、酸はフマル酸である。
【0032】
この反応は、好都合には適切な溶媒、たとえばエーテル類、たとえば2−メチルテトラヒドロフランの存在下で実施される。この反応は、適切には高められた温度、たとえば約70〜105℃、適切には約90℃で実施される。
【0033】
他の態様において、化合物(I)は、式(III)の化合物をN’−(3−クロロ−2−フルオロ−フェニル)−N,N−ジメチル−ホルムアミジン(化合物(XII))と反応させることにより、式(III)の化合物から直接製造できる。この反応は、適切には酸性条件下で実施される。この反応は、適切には、適切な酸、たとえば酢酸、ブタン二酸、プロパン酸、コハク酸、フマル酸およびクエン酸、またはその混合物から選択される1種類以上の酸の存在下で実施される。本発明の1態様において、酸はフマル酸である。
【0034】
式(III)の化合物は、式(IV)の化合物:
【0035】
【化7】
【0036】
の還元を含む方法により製造できる。
【0037】
ニトロ基をアミンに還元するのに適した還元反応は周知である。たとえば、式(IV)の化合物を、適切な還元剤、たとえば亜ジオチン酸ナトリウムの存在下での還元により還元することができる。この反応は適切には水性溶媒、たとえば水性メタノールの存在下で実施される。この反応は、好都合には高められた温度、たとえば40〜60℃で実施される。
【0038】
あるいは、式(IV)の化合物の還元は水素化により、たとえば適切な触媒、たとえばカーボン上パラジウム触媒、たとえばカーボン上10%パラジウム触媒、または白金/バナジウム触媒、たとえばカーボン上1%白金+2%バナジウム触媒を用いる接触水素化により実施できる。この水素化は好都合には水性溶媒、たとえばメタノールまたはアセトニトリルの存在下で実施される。他の態様において、別の溶媒、たとえばメタノール、イソプロパノール、または1:1の比率のメタノール:イソプロパノールの混合物も使用できる。
【0039】
式(IV)の化合物は、式(V)の化合物:
【0040】
【化8】
【0041】
のニトロ化を含む方法により製造できる。
【0042】
式(V)の化合物のニトロ化は、芳香環のニトロ化のための周知の方法を用いて、たとえばそのような反応について周知の条件を用いて本明細書の実施例に示すように、式(V)の化合物を硫酸の存在下に硝酸で処理することにより実施できる。
【0043】
式(V)の化合物は、式(VI)の化合物:
【0044】
【化9】
【0045】
を式(VII)の化合物:
【0046】
【化10】
【0047】
(式中、Lg1は適切な脱離基である)と反応させることを含む方法により製造できる。
【0048】
Lg1により表わされる適切な脱離基には、たとえばハロゲノ、たとえばクロロが含まれる。
【0049】
この反応は、適切には、適切な塩基、たとえばカーボネート、有機アミンまたはアルコキシドの存在下で実施される。具体的な塩基には、たとえば炭酸カリウムまたはトリエタノールアミンが含まれる。
【0050】
この反応は、好都合には、不活性溶媒、たとえばアセトニトリル、またはアルコール類、たとえばエタノールの存在下で実施される。この反応は、適切には、高められた温度、好都合には溶媒の還流温度で実施される。
【0051】
式(VI)の化合物は、たとえば反応スキーム1に示すようにして製造できる:
【0052】
【化11】
【0053】
(スキーム1の脚注)
工程(i):Lg2は適切な脱離基、たとえばハロゲノ、アルカンスルホニルオキシまたはアリールスルホニルオキシ基、たとえばクロロ、ブロモ、メタンスルホニルオキシ、4−ニトロベンゼンスルホニルオキシまたはトルエン−4−スルホニルオキシ基である。適切にはLg2はメタンスルホニルオキシ、4−ニトロベンゼンスルホニルオキシまたはトルエン−4−スルホニルオキシ基であり、たとえばLg2はメタンスルホニルオキシである。
【0054】
Pg1は適切なアミン保護基である。そのような基は、たとえばこの主題に関する多数の一般的テキストのいずれかに記載されているように周知である:たとえば‘Protective
Groups in Organic Synthesis’, Theodora Green著 (出版社: John Wiley & Sons)。アミノ保護基の例には、アシル基、たとえばアルカノイル基、たとえばアセチル、アルコキシカルボニル基、たとえばメトキシカルボニル、エトキシカルボニルもしくはtert−ブトキシカルボニル基、アリールメトキシカルボニル基、たとえばベンジルオキシカルボニル、またはアロイル基、たとえばベンゾイルが含まれる。Pg1の格別な例はtert−ブトキシカルボニルである。
【0055】
この反応は、適切には塩基、たとえばカーボネート、たとえば炭酸カリウムの存在下で実施される。この反応は、好都合には、適切な不活性溶媒、たとえばアルコール類、たとえばイソプロパノールの存在下で実施される。この反応は、適切には、高められた温度、好都合には溶媒の還流温度で実施される。
【0056】
工程(ii):保護基Pg1は常法により除去される。たとえばPg1がtert−ブ
トキシカルボニルである場合、それは塩酸、硫酸もしくはリン酸またはトリフルオロ酢酸など適切な酸で処理することにより除去できる。
【0057】
他の態様において、化合物(I)は、式(III)の化合物を式(XI)の化合物と反応させることにより、式(III)の化合物から直接製造できる。
【0058】
【化12】
【0059】
式(XI)の化合物はN,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミドとも呼ばれる。他の態様において、化合物(I)は、式(III)の化合物を式(XII)の化合物と反応させることにより、式(III)の化合物から直接製造できる。
【0060】
【化13】
【0061】
式(XII)の化合物はN’−(3−クロロ−2−フルオロ−フェニル)−N,N−ジメチル−ホルムアミジンと呼ばれる。これらの反応はいずれも、適切には酸性条件下で実施される。たとえば、この反応は、適切には酢酸、ブタン二酸、フマル酸またはプロパン酸の存在下で実施される。
【0062】
この反応は、好都合には適切な溶媒、たとえばエーテル類、たとえば2−メチルテトラヒドロフラン、またはアルコール類、たとえばエタノールまたはtert−ブチルアルコールの存在下で実施される。この反応は、適切には高められた温度、たとえば約70〜約105℃で実施される。ある態様において、この反応は約80〜約90℃で実施できる。
【0063】
式(XI)の化合物は、3−クロロ−2−フルオロアニリンをオルトギ酸エチルと反応させることにより製造できる。この反応は、適切には酸性条件下で実施される。たとえば、この反応は、適切には酢酸の存在下で実施される。
【0064】
この反応は、好都合には適切な溶媒、たとえばシクロヘキサンの存在下で実施される。この反応は、適切には高められた温度、たとえば約40〜約60℃、適切には約50℃で実施される。
【0065】
したがって、化合物(I)は式(III)の化合物から前記のいずれかの経路で製造できる。
【0066】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0067】
工程1〜2に適切な条件は前記に定めたとおりである。
【0068】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(3a)前記に定めた式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程。
【0069】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(2b)前記に定めた式(III)の化合物を前記に定めた式(XI)の化合物と反応させる工程;および
(1)化合物(I)を単離する工程。
【0070】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(3b)3−クロロ−2−フルオロアニリンをオルトギ酸エチルと反応させて、式(XI)の化合物にする工程。
【0071】
あるいは、本発明の他の観点において、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(2c)前記に定めた式(III)の化合物を前記に定めた式(XII)の化合物と反応させる工程;および
(1)化合物(I)を単離する工程。
【0072】
上記方法はさらに下記の工程を含むことができる:
(3c)3−クロロ−2−フルオロアニリンをN,N−ジメチルホルムアミドジメチルアセタールと反応させて、式(XII)の化合物にする工程。
【0073】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(4)前記に定めた式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程。
【0074】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(5)前記に定めた式(V)の化合物をニトロ化して、前記に定めた式(IV)の化合物にする工程。
【0075】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(6)前記に定めた式(VI)の化合物を前記に定めた式(VII)の化合物と反応させて、前記に定めた式(V)の化合物にする工程。
【0076】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(7)式(VIII)の化合物を脱保護して、前記に定めた式(VI)の化合物にする工程。
【0077】
さらに他の観点において、上記方法はさらに下記の工程を含むことができる:
(8)前記に定めた式(X)の化合物を前記に定めた式(IX)の化合物と反応させて、前記に定めた式(VIII)の化合物にする工程。
【0078】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(3a)前記に定めた式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0079】
工程1〜3に適切な条件は前記に定めたとおりである。
【0080】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(4)前記に定めた式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程;
(3a)式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0081】
工程1〜4に適切な条件は前記に定めたとおりである。
【0082】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(5)前記に定めた式(V)の化合物をニトロ化して、前記に定めた式(IV)の化合物にする工程;
(4)式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程;(3a)式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;そして
(1)化合物(I)を単離する工程。
【0083】
工程1〜5に適切な条件は前記に定めたとおりである。
【0084】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(6)前記に定めた式(VI)の化合物を前記に定めた式(VII)の化合物と反応させて、前記に定めた式(V)の化合物にする工程;
(5)式(V)の化合物をニトロ化して、前記に定めた式(IV)の化合物にする工程;(4)式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程;(3a)式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0085】
工程1〜6に適切な条件は前記に定めたとおりである。
【0086】
本発明の他の観点によれば、下記の工程を含む、化合物(I)またはその医薬的に許容できる塩の製造方法が提供される:
(8)前記に定めた式(X)の化合物を前記に定めた式(IX)の化合物と反応させて、
前記に定めた式(VIII)の化合物にする工程;
(7)式(VIII)の化合物を脱保護して、前記に定めた式(VI)の化合物にする工程;
(6)式(VI)の化合物を前記に定めた式(VII)の化合物と反応させて、前記に定めた式(V)の化合物にする工程;
(5)式(V)の化合物をニトロ化して、前記に定めた式(IV)の化合物にする工程;(4)式(IV)の化合物を還元して、前記に定めた式(III)の化合物にする工程;(3a)式(III)の化合物をN,N−ジメチルホルムアミドジメチルアセタールと反応させて、前記に定めた式(II)の化合物にする工程;
(2a)式(II)の化合物を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させる工程;および
(1)化合物(I)を単離する工程。
【0087】
工程1〜8に適切な条件は前記に定めたとおりである。
【0088】
前記のいずれかの方法において、工程2a)および3a)を下記の工程2b)および3b)で置き換えることができる;
(2b)前記に定めた式(III)の化合物を前記に定めた式(XI)もしくは(XII)の化合物と反応させる工程;および/または
(3b)3−クロロ−2−フルオロアニリンをオルトギ酸エチルと反応させて、式(XI)の化合物にする工程。
【0089】
前記のいずれかの方法において、工程2a)および3a)を下記の工程2c)および3c)で置き換えることができる;
(2c)前記に定めた式(III)の化合物を前記に定めた式(XII)の化合物と反応させる工程;および/または
(3c)3−クロロ−2−フルオロアニリンをN,N−ジメチルホルムアミドジメチルアセタールと反応させて、式(XII)の化合物にする工程。
【0090】
所望により、目的化合物(I)を医薬的に許容できる塩に変換することができる。WO2005/028469に化合物(I)の塩類の例、たとえば化合物(I)と無機酸または有機酸、た
とえば塩酸、臭化水素酸、硫酸、トリフルオロ酢酸、クエン酸またはマレイン酸との酸付加塩が記載されている。格別な塩は実施例に記載するように化合物(I)のジフマレート塩(二フマル酸塩)である。
【0091】
本発明による方法に使用する特定の中間体は新規であり、本発明のさらに他の観点をなす。本発明において提供される中間体またはその塩は下記の構造をもつことができる:
【0092】
【化14】
【0093】
式中:
R1は、H、−NH2、−NO2、または
【0094】
【化15】
【0095】
であり;
R2は、H、N−メチルカルバモイルメチル、またはPg1であり;
Pg1は、アミノ保護基であり;
ただし、R2がHまたはアミノ保護基である場合はR1はHである。
【0096】
したがって、本発明の他の観点は、式(II)、(III)、(IV)、(V)、(VI)および(VIII)のいずれかの化合物、またはその塩から選択される化合物を提供する。本発明の他の観点は、式(XI)の化合物またはその塩を提供する。本発明の他の観点は、式(XII)の化合物またはその塩を提供する。
【0097】
1態様において、たとえば式(VIII)の化合物においてPg1がtert−ブトキシカルボニルである化合物が提供される。
【0098】
式(II)、(XI)および(XII)の化合物を含めて本明細書に記載する化合物は幾何異性中心をもつ場合があり、E−およびZ−異性体として存在する可能性がある。本発明はそのような幾何異性体およびその混合物をすべて包含すると理解すべきである。本発明の1態様において、式(II)の化合物は実質的にE−異性体として存在する。本発明の他の態様において、式(II)の化合物は実質的にZ−異性体として存在する。本発明の1態様において、式(XI)の化合物は実質的にE−異性体として存在する。本発明の他の態様において、式(XI)の化合物は実質的にZ−異性体として存在する。本発明の1態様において、式(XII)の化合物は実質的にE−異性体として存在する。本発明の他の態様において、式(XII)の化合物は実質的にZ−異性体として存在する。
【0099】
これらの中間体は遊離塩基として、または適切な塩の形で使用できる。そのような塩類には、医薬的に許容できる塩類および医薬的に許容できない塩類の両方が含まれる。医薬的に許容できない塩の形の中間体の使用は、本発明による方法に有利な場合がある。たとえば、そのような塩類は中間体の単離または精製のために有用な可能性がある。必要であれば、それらの中間体を常法により修飾して、その化合物の医薬的に許容できる塩にすることができる。そのような方法は当業者に周知であり、たとえば医薬的に許容できる対イオンの存在下での化合物のイオン交換法または再沈殿を含む。したがって、本発明は前記の中間体およびその塩類を含むものであると理解すべきである。
【0100】
化合物(I)およびその塩類、たとえばジフマレート塩は、適切には適切な医薬組成物、たとえば錠剤、カプセル剤または顆粒配合物の形で患者に経口投与される。
【0101】
たとえば化合物(I)のジフマレートは、適切には下記の賦形剤を用いて錠剤として配合される:
錠剤コア:
化合物(I)のジフマレート(たとえば形態A);
乳糖;
微結晶性セルロース;
クロスポビドン;
ポリビドン(PVP);および
ステアリン酸マグネシウム。
【0102】
錠剤コアをフィルムコーティング、たとえばHPMCベースのフィルムコーティングでコートすることができ、このコーティングは場合により1種類以上の着色剤および/または光保護剤を含有する。
【0103】
錠剤は常法により、実施例に示したように製造できる。
【0104】
1種類以上の賦形剤と合わせて単一剤形を製造する有効成分の量は、処置されるホストおよび個々の投与経路に応じて必然的に異なるであろう。たとえば、ヒトに経口投与するための配合物は一般に、たとえば0.5mgから0.5gまでの有効薬剤(より適切には、0.5から200mgまで、たとえば1から100mgまで)を、適切かつ好都合な量の賦形剤と配合して含有するであろう;賦形剤は組成物全体の約5から約98重量%の範囲であってもよい。
【0105】
治療または予防のための化合物(I)およびその塩類、たとえばジフマレート塩の用量サイズは、その状態の性質および重症度、動物または患者の年齢および性別、ならびに投与経路に従い、周知の医薬原理に従って、当然異なるであろう。1態様において、癌、たとえば乳癌の処置に用いるのに適切な化合物(I)の量は、40、80、100、160、200または240mgを1日2回である。
【0106】
化合物(I)は抗増殖特性、たとえば抗癌特性をもち、これはそれらのerbBファミリー受容体型チロシンキナーゼ阻害活性、特に混合erbB2/EGFおよび/またはerbB3/EGFプロフィールから生じると考えられる。
【0107】
したがって、本発明の化合物は、もっぱらまたは部分的にerbB受容体型チロシンキナーゼにより仲介される疾患または病的状態の処置に有用であると予想される。したがって、本発明の化合物は、乾癬、良性前立腺肥大(BPH)、アテローム性硬化症および再狭窄および/または癌の処置に際して、抗増殖作用をもたらすことにより、特にerbB受容体型チロシンキナーゼ感受性癌の処置に有用であると予想される。そのような良性または悪性腫瘍はいかなる組織にも影響を及ぼす可能性があり、これには非充実性腫瘍、たとえば白血病、多発性骨髄腫またはリンパ腫、ならびに充実性腫瘍、たとえば胆管癌、骨癌、膀胱癌、脳/CNS癌、乳癌、結腸直腸癌、子宮内膜癌、胃癌、頭頚部癌、肝癌、肺癌、神経癌、食道癌、卵巣癌、膵臓癌、前立腺癌、腎癌、皮膚癌、精巣癌、甲状腺癌、子宮癌および外陰癌が含まれる。特に、化合物(I)は乳癌の処置に有用であると予想される。
【0108】
化合物(I)およびその塩類、たとえばジフマレートは、エストロゲンおよび/またはプロゲステロン陽性乳癌の処置に際して、有効量のアロマターゼ阻害薬、たとえばアナストロゾール(anastrozole)と組み合わせて使用できる。この組合わせは、以前に内分泌
療法、たとえば選択的エストロゲン受容体調節薬、たとえばタモキシフェン(tamoxifen
)、アロマターゼ阻害薬、たとえばアナストロゾール、またはエストロゲンダウンレギュレーターで処置されていない患者の処置に特に有益な可能性がある。
【0109】
化合物(I)およびその塩類、たとえばジフマレート塩は、タキサン類、たとえばパクリタキセル(paclitaxel)またはドセタキセル(docetaxel)とも組み合わせて使用でき
る。この組合わせは乳癌の処置に有用な可能性がある。たとえば、erbB2の過剰発現が低い乳癌の処置に際して。用語「erbB2の過剰発現が低い(low over-expression of erbB2)」は、Her2の蛍光インサイチュハイブリダイゼーション(fluorescent in-situ hybridization, FISH)が陰性である腫瘍を表わす。「erbB2の過剰発現
が低い」腫瘍である具体的な腫瘍は下記のものである:
(i)免疫組織学化学(immunohistochemistry, IHC)によりHer2 +;および/または
(ii)IHCによりHer2 ++、かつHer2蛍光インサイチュハイブリダイゼーション(FISH)陰性。
【実施例】
【0110】
本発明を以下の実施例によってさらに説明するが、それらは本発明の幾つかの態様を詳述するためのものである。これらの実施例は本発明の範囲を限定するためのものではなく、そのように解釈すべきでもない。ここに具体的に記載したもの以外の形で本発明を実施できることは明らかであろう。本明細書中の教示からみて本発明の多数の改変および変更が可能であり、それらは本発明の範囲に含まれる。
【0111】
実施例中において別途記載しない限り、下記のとおりである。
【0112】
(i)収率は、説明のために示したにすぎず、必ずしも達成可能な最大値ではない。
【0113】
(ii)融点は、Mettler DSC820e装置を用いてDSC分析により測定した。つまり、
1〜2mgの試料を精確に秤量し、ベント付き試料皿内で分析した(加熱を10℃/分で25℃から325℃まで行なった)。別途記載しない限り、本明細書中の融点はDSCを用いて測定した融解吸熱の開始温度を表わす。
【0114】
(iii)質量分析は70電子ボルトの電子エネルギーにより化学イオン化(CI)モードで直接曝露プローブを用いて行なった。指示した場合には、イオン化を電子衝突(EI)、高速原子衝撃(FAB)またはエレクトロスプレー(ESP)により行なった。m/zに関する数値を示す。一般に親質量を指示するイオンのみを報告する。別途記載しない限り、引用した質量イオンは(M+H)+であり、これはプロトン化質量イオンを表わす。M+という表示は電子の喪失により生じた質量イオンに対するものである。(M−H)−という表示はプロトンの喪失により生じた質量イオンに対するものである。
【0115】
(iv)NMRデータを示した場合、それは主診断プロトンについてのデルタ値の形であり、内標準物質としてのテトラメチルシラン(TMS)と対比した百万分率(ppm)で示され、別途指示しない限り、500MHzでペルジュウテリオジメチルスルホキシド(DMSO−d6)を溶媒として用いて測定した。以下の略号を用いた:s,一重線;d,二重線;t,三重線;q,四重線;m,多重線;br(broad),ブロード。
【0116】
(v)化学記号はそれらの通常の意味を有する。SI単位ないし記号を用いる。
【0117】
(vi)溶媒比は体積:体積(v/v)により示される。
【0118】
[実施例1] 4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリン(化合物(I))の製造
化合物(I)を以下に示すスキームに従って製造した。
【0119】
【化16】
【0120】
(工程1) 4−(5−シアノ−2−メトキシフェノキシ)ピペリジン−1−カルボン酸tert−ブチル(中間体2)の製造。
【0121】
3−ヒドロキシ−4−メトキシベンゾニトリル(化合物(X),6.00g,39.62mmole)、(4−メタンスルホニルオキシ)ピペリジン−1−カルボン酸tert−ブチル(16.6g,59.44mmoles)(Chemical & Pharmaceutical Bulletin 2001, 49(7), 822-829)、および炭酸カリウム(6.71g,47.55mmoles
)をイソプロパノール(78.98g)に懸濁し、混合物を撹拌しながら加熱還流した。
さらに(4−メタンスルホニルオキシ)ピペリジン−1−カルボン酸tert−ブチル(2.08g,7.43mmoles)を添加して反応を促進し、完了させた。次いで混合物を水(100.47g)の添加により冷却し、停止させた。中間体2の種晶を添加し、続いて0℃に冷却すると結晶質生成物が生成し、これを濾過により単離した。フィルターケークを水(8.86g)とイソプロパノール(6.97g)の混合物、続いて水(23.64g)で洗浄し、次いで乾燥させて、中間体2を得た(10.75g,80%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 1.48 (m, 2 H) 1.88 (m, 2 H) 3.13 (m, 2 H) 3.67 (m, 2 H) 3.83 (s, 3 H) 4.56 (tt, J=8.1, 3.8 Hz, 1 H) 7.13 (d, J=8.4 Hz, 1 H) 7.42 (dd, J=8.4, 1.9 Hz, 1 H) 7.51 (d, J=1.9 Hz, 1 H); 質量分析 m/z (M + H)+ 333.1。
【0122】
(工程2) 4−メトキシ−3−(ピペリジン−4−イルオキシ)ベンゾニトリル(化合物(VI))の製造。
【0123】
中間体2(39.31g,118.26mmoles)をエタノール(155.53g)に懸濁し、40℃に加熱した。このスラリーにHCl(46.61g,573.04mmoles)を徐々に添加した。混合物を60℃に加熱し、3時間保持した。反応混合物を20℃に冷却し、種晶を装入して結晶化を開始させた。生成した固体を0℃で濾過により単離し、エタノール(62.21g)で2回洗浄し、次いで乾燥させて化合物(VI)を塩酸塩として得た(29.84g,77%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.84 (m, 2 H) 2.09 (m, 2 H) 3.02 (ddd, J=12.7, 8.9, 3.4 Hz, 2 H) 3.20 (m, 2 H) 3.84 (s, 3 H) 4.63 (tt, J=7.7, 3.6 Hz, 1 H) 7.15 (d, J=8.5 Hz, 1 H) 7.45 (dd, J=8.5, 1.9 Hz, 1 H) 7.56 (d, J=1.9 Hz, 1 H) 9.16 (br. s, 2 H); 質量分析 m/z (M +
H)+ 233.2。
【0124】
(工程3) 2−[4−(5−シアノ−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(V))の製造。
【0125】
化合物(VI)(28.36g,95.82mmoles)、2−クロロ−N−メチルアセトアミド(12.37g,114.98mmoles)および炭酸カリウム(33.11g,239.55mmoles)を、アセトニトリル(161.36g)に懸濁した。反応混合物を3時間、加熱還流した。反応混合物を20℃に冷却し、水(386.26g)を装入した。反応物を75℃に加熱し、蒸留により体積を減らした。冷却すると結晶化が起きた。生成した固体を濾過により単離し、水で2回洗浄し(77.25gおよび128.75g)、次いで乾燥させて化合物(V)を得た(27.95g,94%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.68 (m, 2 H) 1.91 (m, 2 H) 2.29 (m, 2 H) 2.61 (d, J=4.7 Hz, 3 H) 2.67 (m, 2 H) 2.88 (s, 2 H) 3.83 (s, 3 H) 4.41 (tt, J=8.3,
4.0 Hz, 1 H) 7.11 (d, J=8.4 Hz, 1 H) 7.40 (dd, J=8.4, 1.9 Hz, 1 H) 7.47 (d, J=1.9 Hz, 1 H) 7.68 (q, J=4.7 Hz, 1 H); 質量分析 m/z (M + H)+ 304.2。
【0126】
(工程4) 2−[4−(5−シアノ−2−メトキシ−4−ニトロフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(IV))の製造。
【0127】
化合物(V)(8.78g,26.11mmoles)を酢酸(22.82g,364.87mmoles)に懸濁し、得られた反応混合物を5℃に冷却した。これに、温度を30℃より低く維持しながら硫酸(23.64g,234.95mmoles)を添加した。得られた溶液に硝酸(2.40g,26.63mmoles)を添加した。次いで反応混合物を35℃に加熱し、3時間保持した。さらに硝酸(117mg,1.31mmoles)および硫酸(1.31g,13.1mmoles)を装入し、反応混合物を35℃で30分間加熱した。この溶液を20℃に冷却し、アンモニア水(92.45g,1.
36moles)で停止させると、温度が50℃に上昇した。得られたスラリーに、プロピオニトリル(61.58g,1.12moles)および水(19g)を添加した。反応混合物を80℃に加熱すると透明な溶液が得られ、これは沈降すると2層になった。下層を分離した。反応混合物を20℃に冷却すると濃厚なスラリーが得られた。固体を濾過により単離し、プロピオニトリル(6.16g,112.0mmoles)で洗浄し、乾燥させて化合物(IV)を得た(7.44g,82%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.72 (m, 2 H) 1.97 (m, 2 H) 2.35 (m, 2 H) 2.61 (d, J=4.7 Hz, 3 H) 2.66 (m, 2 H) 2.90 (s, 2 H) 3.96 (s, 3 H) 4.73 (tt, J=8.4, 4.0 Hz, 1 H) 7.71 (q, J=4.7 Hz, 1 H) 7.82 (s, 1 H) 7.86 (s, 1 H). 質量分析 m/z (M + H)+ 349.2。
【0128】
(工程5) 2−[4−(4−アミノ−5−シアノ−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(III))の製造。
【0129】
化合物(IV)(7.42g,19.38mmoles)を水(44.52g)およびメタノール(5.35g)に懸濁した。これに亜ジオチン酸ナトリウム(11.91g,58.15mmoles)を添加し、得られた反応混合物を60℃に加熱した。反応混合物に塩酸(46.98g,463.89mmoles))を添加すると溶液が得られ、これを60℃に3時間保持した。次いで反応混合物を20℃にまで放冷した。水酸化ナトリウム水溶液(15.51g,182.2mmoles)、続いて2−メチルテトラヒドロフラン(58.0g)を装入した。反応混合物を60℃に加熱し、これは沈降すると2層になり、下層を廃棄した。反応混合物の体積を真空蒸留により減らし、メチル tert−ブチルエーテル(18.54g)を添加するとスラリーが得られ、これを10℃に冷却し、次いで固体を濾過により採集した。固体を2−メチルテトラヒドロフラン(5.8g)で洗浄し、乾燥させて化合物(III)を得た(5.4g,78%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.62 (m, 2 H) 1.82 (m, 2 H) 2.20 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.65 (m, 2 H) 2.86 (s, 2 H) 3.72 (s, 3 H) 4.00 (tt, J=8.3, 4.0 Hz, 1 H) 5.66 (br. s, 2 H) 6.39 (s, 1 H) 6.94 (s, 1 H) 7.65 (q, J=4.7 Hz, 1 H). 質量分析 m/z (M + H)+ 319.2。
【0130】
(工程6) 2−[4−(5−シアノ−4−{[(ジメチルアミノ)メチレン]アミノ}−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(II))の製造。
【0131】
化合物(III)(18.21g,52.05mmoles)を2−メチルテトラヒドロフラン(99.62g)に懸濁した。これに酢酸(162.79mg)およびN,N−ジメチルホルムアミドジメチルアセタール(DMA)(8.63g,70.27mmoles)を添加し、得られた反応混合物を76℃で16時間加熱した。さらにN,N−ジメチルホルムアミドジメチルアセタール(639.41mg,5.20mmoles)を反応混合物に添加して、反応を確実に完了させた。反応混合物を30℃に冷却すると、その間に結晶化が起きた。生成した固体を濾過により単離し、2−メチルテトラヒドロフラン(14.23g)で洗浄して、化合物(II)を得た(19.53g,97%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.65 (m, 2 H) 1.86 (m, 2 H) 2.24 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.66 (m, 2 H) 2.87 (s, 2 H) 2.95 (s, 3 H) 3.04 (s, 3 H) 3.81 (s, 3 H) 4.19 (tt, J=8.2, 3.8 Hz, 1 H) 6.72 (s, 1 H) 7.15 (s, 1 H) 7.67 (q, J=4.7 Hz, 1 H) 7.90 (s, 1 H); 質量分析 m/z (M + H)+ 374.2。
【0132】
(工程7) 化合物(I)の製造。
【0133】
2−[4−(5−シアノ−4−{[(ジメチルアミノ)メチレン]アミノ}−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(II),
7.00g,17.71mmoles)をメトキシベンゼン(35.8g)に懸濁した。酢酸(16.6g)を装入し、得られた溶液に3−クロロ−2−フルオロアニリン(2.71g,18.07mmoles)を添加した。反応混合物を90℃で20時間加熱し、次いで20℃に冷却した。水(37.04g)を反応混合物に装入し、有機層を廃棄した。得られた水性混合物にイソプロパノール(39.00g)、続いてアンモニア水(20.79g,25%)を装入した。反応混合物を30℃に加熱し、化合物(I)の種晶を添加すると、これにより結晶化が誘発された。次いで反応物を0℃に冷却し、生成物を濾過により単離した。フィルターケークを水(7.28g)とイソプロパノール(4.68g)の混合物で2回洗浄し、次いで乾燥させて化合物(I)を得た(5.65g,55%の収率); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (m, 2 H) 2.04 (m, 2 H) 2.38 (m, 2 H) 2.62 (d, J=4.5 Hz, 3 H) 2.74 (m, 2 H) 2.94 (s, 2 H) 3.93 (s, 3 H) 4.56 (tt, J=8.1, 3.8 Hz, 1 H) 7.21 (s, 1 H) 7.28 (m, 1 H) 7.50 (m, 2 H) 7.73 (q, J=4.5 Hz, 1 H) 7.81 (s, 1 H) 8.36 (s, 1 H) 9.56 (br.s, 1 H); 質量分析 m/z (M + H)+ 474.2, 476.2。
【0134】
[実施例2] 4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリン(化合物(I))の製造
化合物(I)を以下に示すスキームに従って製造した。
【0135】
【化17】
【0136】
工程1、2、3および4は実施例1に示したとおりである。
【0137】
(工程5,別法1) 化合物(III)の製造。
【0138】
2−[4−(5−シアノ−2−メトキシ−4−ニトロフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(IV),15.00g,42.50mmoles)を水(90.00g)およびメタノール(59.38g)に懸濁した。これに亜ジオチン酸ナトリウム(30.47g,148.75mmoles)および水(90.00g)を添加し、得られた反応混合物を30℃に加熱し、2時間保持した。反応混合物に塩酸(27.98g,276.25mmoles))を添加すると溶液が得られ、これを60℃に2時間保持した。水酸化ナトリウム水溶液(30.60g,382.49mmoles)、続いてライン洗浄(line wash)水(30.00g)を添加した。反応混合物を2
5℃に冷却するとスラリーが得られ、これを濾過により採集した。固体を水(30.00g)で洗浄し、乾燥させて化合物(III)を得た(13.50g,82%の収率); 1H
NMR (400 MHz, DMSO-d6) δ ppm 1.62 (m, 2 H) 1.82 (m, 2 H) 2.20 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.65 (m, 2 H) 2.86 (s, 2 H) 3.72 (s, 3 H) 4.00 (tt, J=8.3, 4.0 Hz, 1 H) 5.66 (br. s, 2 H) 6.39 (s, 1 H) 6.94 (s, 1 H) 7.65 (q, J=4.7 Hz, 1 H). 質量分析 m/z (M+H)+ 319.2。
【0139】
(工程5,別法2) 化合物(III)の製造。
【0140】
化合物(IV)(8.00g,22.67mmoles)およびカーボン上1%白金+2%バナジウム触媒(1.23g,0.023mmoles)をアセトニトリル(94.00g)に懸濁した。反応混合物を3Bar Gの圧力で35℃の温度において3時間水素化した。完了した時点で反応混合物を濾過して触媒を分離し、アセトニトリル(31.33g)で洗浄した。反応混合物の体積を真空蒸留により減らすとスラリーが得られ、これを0℃に冷却し、次いで固体を濾過により採集した。固体をアセトニトリル(12.53g)で洗浄し、乾燥させて化合物(III)を得た(5.88g,78%の収率); 1H
NMR (400 MHz, DMSO-d6) δ ppm 1.62 (m, 2 H) 1.82 (m, 2 H) 2.20 (m, 2 H) 2.60 (d, J=4.7 Hz, 3 H) 2.65 (m, 2 H) 2.86 (s, 2 H) 3.72 (s, 3 H) 4.00 (tt, J=8.3, 4.0 Hz, 1 H) 5.66 (br. s, 2 H) 6.39 (s, 1 H) 6.94 (s, 1 H) 7.65 (q, J=4.7 Hz, 1 H). 質量分析 m/z (M+H)+ 319.2。
【0141】
(工程6) N,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミド(化合物(XI))の製造。
【0142】
3−クロロ−2−フルロアニリン(51.21g,341.22mmoles)をシクロヘキサン(87.07g)に懸濁した。これにオルトギ酸エチル(22.28g,150.32mmoles)および酢酸(0.94g,15.03mmoles)を添加した。得られた反応混合物を撹拌しながら48℃に12時間加熱した。これに続いて反応混合物を12時間かけて20℃に冷却し、固体生成物を濾過により単離した。フィルターケークをシクロヘキサン(26.12g)で洗浄し、40℃で真空乾燥して、化合物(XI)を白色結晶質生成物として得た(33.95g,93%の収率); 1H NMRスペクトル (400 MHz, DMSO-d6) δ ppm 7.14 (t, 2 H) 7.22 (m, 2 H) 8.14 (s, 1 H), 9,98 (s, 1 H);
質量分析(by GC-MS EI): m/z (M+) 300.0。
【0143】
(工程7,別法1) 化合物(I)の製造。
【0144】
2−[4−(4−アミノ−5−シアノ−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(III))(10g,29.84mmol)およびN,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミド(化合物(XI))(11.46g,37.3mmol)を2−メチルテトラヒドロフラン(30.4ml)に懸濁し、80℃に加熱した。この黄色懸濁液に酢酸(7.6ml,127.33mmol)を添加し、得られた溶液を92℃に6時間加熱した。2−メチルテトラヒ
ドロフラン(66.5ml)および水(28.5ml)を添加し、混合物を55℃に冷却した後、50% w/w水酸化ナトリウム(7ml,131.29mmol)を添加すると温度が63℃に上昇した。温度をさらに69℃に高め、沈降後に水層を廃棄した。有機層を水で洗浄し(3×20ml)、それぞれ水層を沈降後に廃棄した。2−メチルテトラヒドロフラン(100ml,997mmol)を添加し、蒸留により体積を減らした。種晶を添加して結晶化を誘発し、得られた混合物を15℃に冷却した。上記実験から自然結晶化に伴って結晶質形態が最初に得られた。生成した固体を濾過により単離し、2−メチルテトラヒドロフラン(19ml)で2回洗浄し、40℃で真空乾燥して、化合物(I)を白色固体として得た(12.14g,95%); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.12 (d, J= 6Hz,1.3H), 1.26 -1.36 (m, 0.4H), 1.75-1.97 (m, 3.3H), 2.02-2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.7Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H),
3.52-3.59 (m, 0.4H), 3.72-3.87 (m, 0.86H), 3.95 (s, 3H), 4.53-4.63 (m, 1H), 7.22 (s, 1H), 7.29 (dt J= 1Hz J= 8Hz, 1H), 7.51 (dt J= 7.4Hz, J= 18Hz, 2H), 7.71-7.77 (m, 1H), 7.82 (s, 1H), 8.37 (s, 1H), 9.57 (s, 1H). 質量分析 m/z (M+H)+ 474.0
。上記のNMRデータは0.43モル当量で存在する2−メチルテトラヒドロフラン溶媒に関する信号をも含む。溶媒に関係する信号はδ ppmシフト1.12, 1.26-1.36, 3.52-3.59および3.72-3.87にあるものである。1.75-1.93にあるクラスターは溶媒と親化合物に関する信号を含む。この化合物のXRPDを図2に示す。
【0145】
(工程7,別法2) 化合物(I)の製造。
【0146】
化合物(III)(15g,44.76mmol)および化合物(XI)(17.19g,55.95mmol)を2−メチルテトラヒドロフラン(45.6ml)に懸濁し、83℃に加熱した。この黄色懸濁液に酢酸(11.4ml,190.99mmol)を添加し、得られた溶液を92℃に3.5時間加熱した。2−メチルテトラヒドロフラン(105ml)および水(50ml)を添加し、混合物を49℃に冷却した後、50% w/w水酸化ナトリウム(10.74ml,201.4mmol)を添加すると温度が62℃に上昇した。温度を62℃に保持し、沈降後に水層を廃棄した。有機層を水で洗浄し(3×30ml)、沈降後にそれぞれ水層を廃棄した。混合物を15℃に冷却し、種晶を添加して結晶化を誘発した。上記実験から自然結晶化に伴って結晶質形態が最初に得られた。生成した固体を濾過により単離し、2−メチルテトラヒドロフラン(21ml)で2回洗浄し、40℃で真空乾燥して、化合物(I)を白色固体として得た(20.12g,95%). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.75-1.86 (m, 2H), 2.02-2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.7Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.95 (s, 3H), 4.53-4.63 (m, 1H), 7.22 (s, 1H), 7.29 (dt J= 1Hz J= 8Hz, 1H), 7.51 (dt J=
7.4Hz, J= 18Hz, 2H), 7.71-7.77 (m, 1H), 7.82 (s, 1H), 8.37 (s, 1H), 9.57 (s, 1H). 質量分析 m/z (M+H)+ 474.0。この化合物のXRPDを図3に示す。
【0147】
(工程7,別法3) 化合物(I)の製造。
【0148】
化合物(III)(15.1g,45.06mmol)および化合物(XI)(17.31g,56.32mmol)を2−メチルテトラヒドロフラン(46ml)に懸濁し、80℃に加熱した。この黄色懸濁液に酢酸(12ml,458mmol)を添加し、得られた溶液を92℃に7時間加熱した。2−メチルテトラヒドロフラン(100ml)および水(43ml)を添加し、混合物を59℃に冷却した後、50% w/w水酸化ナトリウム(11ml,207mmol)を添加すると温度が71.5℃に上昇した。温度を69℃に調整し、沈降後に水層を廃棄した。有機層を水で洗浄し(2×43ml)、沈降後にそれぞれ水層を廃棄した。2−メチルテトラヒドロフラン(72ml)を大気圧蒸留により除去し、イソプロピルアルコール(72ml)の添加により交換した。さらに72mlの溶媒を大気圧蒸留により除去し、イソプロピルアルコール(72ml)により交換し
た。種晶を添加して結晶化を誘発し、得られた混合物を15℃に冷却した。固体を濾過により単離し、イソプロピルアルコール(32ml)で2回洗浄し、40℃で真空乾燥して、化合物(I)を白色固体として得た(20.86g,87%). 1H NMR (400 MHz, DMSO-d6)δ ppm 1.04 (d, J= 6Hz, 6H),1.75-1.88 (m, 2H), 2.02-2.15 (m, 2H), 2.35-2.44
(m, 2H), 2.64 (d, J= 4.7Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.73-3.84 (m,
1H), 3.95 (s, 3H), 4.34 (d, J = 4.2Hz, 1H), 4.53-4.63 (m, 1H), 7.22 (s, 1H), 7.29 (dt J= 1Hz J= 8Hz, 1H), 7.51 (dt J= 7Hz, J= 18Hz, 2H), 7.71-7.77 (m, 1H), 7.82 (s, 1H), 8.37 (s, 1H), 9.57 (s, 1H). 質量分析 m/z (M+H)+ 474.0。NMRデータは存在する1モル当量のイソプロパノールに関する信号を含む。この化合物のXRPDを図4に示す。
【0149】
[実施例3] 4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリン ジ−[(2E)−ブタ−2−エンジオエート](化合物(I)ジフマレート塩)の製造
化合物(I)ジフマレート塩を以下に示すスキームに従って製造した。
【0150】
【化18】
【0151】
工程1、2、3、4、5および6は実施例2に示したとおり実施した。
【0152】
(工程7) 化合物(I)ジフマレート塩の製造。
【0153】
化合物(III)(17.90mmoles)およびN,N’−ビス(3−クロロ−2−フルオロフェニル)イミドホルムアミド(化合物(XI))(7.04g,23.27mmoles)をtert−ブチルアルコール(88.95g)に懸濁した。この懸濁液にフマル酸(10.39g,89.52mmoles)を添加し、混合物を撹拌しながら80℃に2.5時間加熱した。水(11.40g,632.80mmoles)を装入し、反応をさらに21.5時間続けた。反応物を12時間かけて20℃にまで冷却すると、その間に結晶化が起きた。生成した固体を濾過により単離し、水(1.00)とtert−ブチルアルコール(7.80g)の混合物で洗浄し、続いて水(0.50g)とtert−ブチルアルコール(7.30g)の混合物で洗浄した。固体を40℃で真空乾燥して、化合物(I)ジフマレート塩(8.17g,61.40%)をマスタード黄色粉末として得た; 1H NMR (400 MHz, DMSO-d6) δ ppm 1.83 (m, 2 H, broad) 2.07 (m, 2 H, broad) 2.64 (d, J=5.0 Hz, 3 H) 2.80 (m, 2 H, broad) 3.03 (s, 2 H) 3.94 (s, 3 H) 4.58
(m, 1 H) 6.63 (s, 4 H) 7.22 (s, 1 H) 7.29 (td, J=8.5, 1.0 Hz, 1 H) 7.51 (m, 2 H) 7.82 (m, 2 H) 8.37 (s, 1 H); 質量分析 m/z (M+H)+ 474.0。
【0154】
[実施例4] 4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリン(化合物(I))の製造
化合物(I)を以下に示すスキームに従って製造した。
【0155】
【化19】
【0156】
工程1、2、3、4および5は実施例2に示したとおり実施した。
【0157】
(工程6) N’−(3−クロロ−2−フルオロ−フェニル)−N,N−ジメチル−ホルムアミジン(化合物(XII))の製造。
【0158】
3−クロロ−2−フルロアニリン(5.30g,35.29mmoles)を2−メチルテトラヒドロフラン(52.94g)に溶解した。これにN,N−ジメチルホルムアミドジメチルアセタール(6.07g,49.41mmoles)および酢酸(0.11g,1.76mmoles)を添加した。得られた反応混合物を撹拌しながら76℃に3時間加熱した。これに続いて溶液を40℃で真空下に除去して、化合物(XII)を黄色の油として得た(6.60g,93%の収率); 1H NMRスペクトル (400 MHz, DMSO-d6) δ
ppm 2.74 (s, 0.29H), 2.89 (s, 0.31H), 2.94 (s, 2.75H), 3.03 (s, 2.66H), 3.34 (br s, 0.70H), 5.48 (s, 0.06H) 6.91-7.10 (m, 3H), 7.79 (s, 1 H), 7.96 (s, 1 H)。上記のNMRデータは0.06モル当量で存在するN,N−ジメチルホルムアミドジメチルアセタールに関する信号を含む。N,N−ジメチルホルムアミドジメチルアセタールに関係する信号はδ ppmシフト3.75、および6.90-6.95にあるものである。δ ppm 3.35にある信号は残留水によるものである。質量分析(LCMS EIによる): m/z (M+H)+ 201.2。
【0159】
(工程7) 化合物(I)の製造。
【0160】
2−[4−(4−アミノ−5−シアノ−2−メトキシフェノキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(III))(0.50g,1.45mmol)およびN’−(3−クロロ−2−フルオロ−フェニル)−N,N−ジメチル−ホルムアミジン(化合物(XII))(0.32g,1.52mmol)を、メトキシベンゼン(3.1ml)に懸濁した。この黄色懸濁液に酢酸(1.52ml,25.51mmol)を添加し、得られた溶液を90℃に14時間加熱した。反応混合物を20℃に冷却し、水(2.58mL)を添加した。有機層を除去し、水層をメトキシベンゼン(1.4mL)で洗浄した。エタノール(2.45mL)およびアンモニア(1.94ml,25.55mmoles)を水層に添加した。この溶液を90℃に加熱すると、若干のエタノールが蒸発により失われた。この溶液を40℃に冷却した。種晶を添加して結晶化を誘発し、得られた混合物を20℃に冷却した。固体を濾過により単離して、化合物(I)を白色固体として得た(0.61g,73%の収率). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.75-1.87
(m, 2H), 2.02-2.15 (m, 2H), 2.35-2.44 (m, 2H), 2.64 (d, J= 4.8Hz, 3H), 2.72-2.80 (m, 2H), 2.95 (s, 2H), 3.35 (s, 5.4H), 3.75 (s, 1.3H), 3.95 (s, 3H), 4.58 (hept., J=4.0Hz, 1H), 6.90-6.95 (m, 1.3H), 7.23 (s, 1.8H), 7.26-7.34 (m, 1H), 7.45-7.58 (m 2H), 7.72-7.78 (m, 1H), 7.83 (s, 1H), 8.38 (s, 1H), 9.58 (s, 1H)。上記の
NMRデータは0.40モル当量で存在するメトキシベンゼン溶媒に関する信号を含む。この溶媒に関係する信号はδ ppmシフト3.75、および6.90-6.95にあるものである。7.26-7.34にあるクラスターは溶媒と親化合物に関する信号を含む。δ ppm 3.35にある信号は
残留水によるものである。質量分析 m/z (M + H)+ 474.0, 476.0。
【0161】
[実施例5] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエート]形態Aの製造
フマル酸(2.7g,23.22mmol)のメタノール(95ml)中における溶液を、2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(I))(5.62g,89% w/wのもの,10.55mmol)のイソプロパノール(100ml)中における混合物に、温度を>65℃に維持しながら添加した。混合物を澄明になるまで1時間、加熱還流した。反応混合物を90分間かけて30℃にまで冷却し、30分間保持して結晶化を確立した。反応物を2時間かけて0℃にまで冷却し、1時間保持した後、濾過により単離した。フィルターケークを冷イソプロパノールで2
回洗浄し(2×10ml)、50℃で真空乾燥して、表題化合物を白色固体として得た(5.84g,78%); 1H NMRスペクトル: (DMSO) 1.85 (m, 1H), 2.08 (m, 1H), 2.50 (m, 1H), 2.66 (d, 3H), 2.83 (m, 1H), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, 1H), 6.64 (s, 4H), 7.23 (s, 1H), 7.28 (m, 1H), 7.46 (ddd, 1H), 7.55 (m, 1H), 7.70 (broad q, 1H), 7.85 (s, 1H), 8.38 (s, 1H)。
【0162】
[実施例6] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエート]形態Aの製造
フマル酸(1.4kg,12.1mol)のメタノール(26.6kg)中における溶液を、2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(2.93kg,84.8% w/w,5.24mol)のイソプロパノール(39kg)中における混合物に、温度を>65℃に維持しながら添加した。ライン洗浄メタノール(3.6kg)を装入した。混合物を澄明になるまで1時間、加熱還流し、続いてライン洗浄メタノール(7kg)を添加した。反応混合物を大気圧蒸留して47kgの留出物を除去した。イソプロパノール(15.8kgを添加し、反応混合物を蒸留して15.6kgの留出物を除去した。蒸留中に結晶化が起きた。イソプロパノール(21kg)を添加し、反応物を8時間かけて0℃に冷却し、1時間保持した後、濾過により単離した。フィルターケークを、冷却した50:50イソプロパノール:MeOH(4kg)、続いて冷イソプロパノール(4kg)で洗浄し、50℃で真空乾燥して、表題化合物を白色固体として得た(3.64kg,98%); 1H NMRスペクトル: (DMSO) 1.85 (m, 1H), 2.08 (m, 1H), 2.50 (m, 1H), 2.66 (d, 3H), 2.83 (m, 1H), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, 1H), 6.64 (s, 4H), 7.23 (s, 1H), 7.28 (m, 1H), 7.46 (ddd, 1H), 7.55 (m, 1H), 7.70 (broad q, 1H), 7.85 (s, 1H), 8.38 (s, 1H)。
【0163】
[実施例7] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエート]形態Aの製造
2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(I))(60.19g,88% w/wのもの,111.8mmol)を酢酸エチル(1550ml)に溶解した。この溶液を濾過により澄明化し、フィルターを酢酸エチル(53ml)で洗浄した。この溶液を40℃に冷却した。次いで、フマル酸(26.60g,257.0mmol)のイソプロパノール(408ml)中における澄明化した溶液を1時間かけて添加した。フマル酸溶液を澄明化するために用いたフィルターを、次いでイソプロパノール(37ml)で洗浄した。40℃に1時間保持した後、反応物を1時間かけて20℃にまで冷却した。反応混合物を13.5時間保持した後、生成物を濾過により単離した。フィルターケークを酢酸エチル(82ml):イソプロパノール(24ml)で2回洗浄し、次いで40℃で真空乾燥して、表題化合物を白色固体として得た(72.32g,90%); 1H NMRスペクトル: (DMSO) 1.85 (m, 1H), 2.08 (m, 1H), 2.50 (m, 1H), 2.66 (d, 3H), 2.83 (m, 1H), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, 1H), 6.64 (s, 4H), 7.23 (s, 1H), 7.28 (m, 1H), 7.46 (ddd, 1H), 7.55 (m, 1H), 7.70 (broad q, 1H), 7.85 (s, 1H), 8.38 (s, 1H)。
【0164】
[実施例8] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエ
ート]形態Aの製造
2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(I))(2.75g,推定100% w/wのもの,5.80mmol)を、酢酸エチル(94ml)およびイソプロパノール(14ml)に溶解した。溶液を蒸留し、これにより25.2mlの留出物を採集した。溶液を40℃に冷却した。フマル酸(1.38g,11.90mmol)のイソプロパノール(21ml)中における澄明化した溶液を、次いで1時間かけて添加した。化合物(I)ジフマレート形態Aの種晶(3.7mg,5.3μmol)を添加した。フマル酸溶液を澄明化するために用いたフィルターを、次いでイソプロパノール(2ml)で洗浄した。40℃に1時間保持した後、反応物を2時間かけて20℃にまで冷却した。反応混合物を15時間保持した後、生成物を濾過により単離した。フィルターケークを酢酸エチル(4.3ml):イソプロパノール(1.2ml)で2回洗浄し、次いで40℃で真空乾燥して、表題化合物を白色固体として得た(72.32g,90%); 1H NMRスペクトル: (DMSO) 1.85 (m, 1H), 2.08 (m, 1H), 2.50 (m, 1H), 2.66 (d, 3H), 2.83 (m, 1H), 3.05 (s, 2H), 3.96 (s, 3H), 4.58 (m, 1H),
6.64 (s, 4H), 7.23 (s, 1H), 7.28 (m, 1H), 7.46 (ddd, 1H), 7.55 (m, 1H), 7.70 (broad q, 1H), 7.85 (s, 1H), 8.38 (s, 1H)。
【0165】
[実施例9] 化合物(I)ジフマレート形態A:2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド ジ−[(2E)−ブタ−2−エンジオエート]形態Aの製造
2−[4−({4−[(3−クロロ−2−フルオロフェニル)アミノ]−7−メトキシキナゾリン−6−イル}オキシ)ピペリジン−1−イル]−N−メチルアセトアミド(化合物(I))(1g,1.86mmoles)およびフマル酸(0.44g,3.81mmoles)を水(4.4g)に懸濁し、85℃に加熱した。反応混合物を1℃/分で60℃にまで冷却し、温度が77℃になった時点で化合物(I)形態Aの種晶を添加した。生成した固体を濾過により単離し、アセトンで2回洗浄し(1回の洗浄当たり0.70g)、40℃で真空乾燥して、表題化合物を得た(0.89g,68%の収率); 1H NMR (400 MHz, DMSO-d6) d ppm 1.84 (m, 2 H) 2.08 (m, 2 H) 2.55 (m, 2 H) 2.63 (d, J=4.7
Hz, 3 H) 2.86 (m, 2 H) 3.12 (s, 2 H) 3.93 (s, 3 H) 4.59 (tt, J=7.8, 3.7 Hz, 1 H) 6.62 (s, 4 H) 7.21 (s, 1 H) 7.27 (td, J=8.1, 1.3 Hz, 1 H) 7.49 (m, 2 H) 7.86 (m, 2 H) 8.36 (s, 1 H) 9.63 (br. s., 1 H)。
【0166】
化合物(I)ジフマレート形態Aはさらさらした粉末である。化合物(I)ジフマレートのX線粉末回折(図1)は、この物質が結晶質であることを示す。X線粉末回折分析は、シンチレーション検出器を備えたSiemens D5000粉末X線回折計を用いて実施した;X
線源は、波長1.54Åを生じるCu Kαであった;データを2−θ範囲2〜40°にわたって2−θ 0.02°の増分で、増分当たり1秒を用いて収集し、次表に同定するカテゴリーに分類した:
【0167】
【表1】
【0168】
X線粉末回折の技術分野の専門家には、ピークの相対強度が、たとえば試料の分析に影響を及ぼす30ミクロンを超えるサイズの結晶粒および不均一なアスペクト比により影響される可能性があることが分かるであろう。反射の位置は試料が回折計内で置かれた厳密な高さおよび回折計のゼロ目盛定めにより影響される可能性があることも当業者には分かるであろう。試料の表面平面性もわずかな影響をもつ可能性がある。したがって、提示した回折パターンデータは絶対値と解釈すべきではない(詳細な情報については、Jenkins, R & Snyder, R.L. ‘Introduction to X-Ray Powder Diffractometry’ John Wiley & Sons, 1996を参照)。
【0169】
[実施例10] 化合物(I)ジフマレートの錠剤配合物
粉末成分をミキサーに装入し、混合して薬物の均一な分散物を得る。結合剤溶液を調製し、粉末に添加し、適切な湿潤素材が形成されるまでさらに混合する。湿潤素材をスクリーンに通し、得られた顆粒を乾燥させて適宜な含水率にする。乾燥顆粒を適宜なサイズのスクリーンに通し、そしてステアリン酸マグネシウムとブレンドした後、一般的な錠剤製造装置により圧縮して錠剤コアにする。圧縮したコアを、一般的な有孔ドラムコーターによりフィルムコーティング成分の水性懸濁液でコートする。
【0170】
上記に従って製造した、2.5、10、40および100mgの化合物(I)に相当する化合物(I)ジフマレート形態Aを含有するフィルムコートした錠剤を、下記の表1に示す。
【0171】
【表2】
【0172】
錠剤を製造するのに適した製造方法を下記に概説する。
【0173】
【表3】
【0174】
本明細書に記載する化合物は遊離の非塩形で生じる可能性があり、あるいは塩類として生じる可能性があり、用語「化合物」の使用が遊離形の化合物および化合物の塩類を包含することは当業者には認識されるであろう。
【0175】
当業者に理解されるように、いずれかまたはすべての目的について、特に記述提示に関して、本明細書に開示するすべての範囲は、そのすべての可能な下位範囲および下位範囲の組合わせ、ならびにその範囲を構成する個々の数値、特に整数値をも包含する。列記した範囲はいずれも、その範囲が少なくとも2、3、4、5、6等分などされることを十分に記述および可能にすることは、容易に認識できる。限定ではない例として、本明細書中に述べる各範囲は、下1/3、中1/3、上1/3などに容易に分割できる。同様に当業者に理解されるように、「最高で(up to)」、「少なくとも(at least)」、「より大
きい(greater than)」、「より少ない(less than)」、「より多い(more than)」、「またはそれより多い(or more)」などの語はすべて、引用した数値を含み、さらに前
記に述べたように下位範囲に分割できる範囲を表わす。同様に、本明細書に開示するすべての比率は、その広い比率内に含まれるすべての下位比率をも含む。
【0176】
たとえばMarkushグループのようにメンバーを一般的な方法でグループにまとめた場合
、本発明はまとめて挙げたグループ全体だけでなくそのグループの各メンバー個々および
その主グループの可能なすべての下位グループをも包含することも、当業者には容易に認識されるであろう。さらに、すべての目的に関して、本発明は主グループだけでなく1以上のグループメンバーが存在しない主グループをも包含する。本発明は本発明内のいずれか1以上のグループメンバーの明らかな除外をも考慮する。
【0177】
当業者に理解されるように、成分量、特性、たとえば分子量、反応条件などを表わすものを含めたすべての数値は概数であり、すべての例において用語「約」で修飾されていると理解される。これらの数値は、本発明の教示を利用して当業者が求める目的特性に応じて変動する可能性がある。そのような数値は本来、それらのそれぞれの試験測定値にみられる標準偏差から必然的に生じる変動性を含むことも理解される。
【0178】
本明細書に開示するすべての参考文献を、本明細書に具体的に援用する。
【0179】
本明細書中の「工程」という表記、または「工程」に付けた番号は便宜的に用いたものにすぎず、本明細書中に述べる本発明を分類、規定または限定しない。
【0180】
特定の態様を説明および記載したが、これらの態様は本発明の範囲を限定しないこと、ならびに特許請求の範囲に定めた本発明の広い観点から逸脱することなく当業者が変更および改変しうることを理解すべきである。
【特許請求の範囲】
【請求項1】
4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリンまたはその塩の製造方法であって:
(a)式(II)の化合物:
【化1】
を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させること;または
(b)式(III)の化合物:
【化2】
を適切な酸の存在下で式(XI)または式(XII)の化合物:
【化3】
と反応させること;
を含む方法。
【請求項2】
酸が酢酸、ブタン二酸、フマル酸またはプロパン酸である、請求項1に記載の方法。
【請求項3】
式(II)の化合物が、式(III)の化合物:
【化4】
をN,N−ジメチルホルムアミドジメチルアセタールと反応させることを含む方法により製造される、請求項1に記載の方法。
【請求項4】
式(III)の化合物が、式(IV)の化合物:
【化5】
の還元を含む方法により製造される、請求項1または3に記載の方法。
【請求項5】
式(IV)の化合物が、式(V)の化合物:
【化6】
のニトロ化を含む方法により製造される、請求項4に記載の方法。
【請求項6】
式(V)の化合物が、式(VI)の化合物:
【化7】
を式(VII)の化合物:
【化8】
(式中、Lg1は適切な脱離基である)と反応させることを含む方法により製造される、請求項5に記載の方法。
【請求項7】
式(VI)の化合物が、式(VIII)の化合物:
【化9】
(式中、Pg1は適切なアミノ保護基である)の脱保護を含む方法により製造される、請求項6に記載の方法。
【請求項8】
式(VIII)の化合物が、式(X)の化合物:
【化10】
を式(IX)の化合物:
【化11】
(式中、Lg2は適切な脱離基であり、Pg1は適切なアミノ保護基である)と反応させることを含む方法により製造される、請求項7に記載の方法。
【請求項9】
式(XIII)の化合物またはその塩:
【化12】
(式中、R1は、H、−NH2、−NO2、または
【化13】
であり;
R2は、H、N−メチルカルバモイルメチル、またはPg1であり;
Pg1は、アミノ保護基であり;
ただし、R2がHまたはアミノ保護基である場合はR1はHである)。
【請求項10】
式(II)、(III)、(IV)、(V)、(VI)および(VIII)のいずれかの化合物から選択される、請求項9に記載の化合物:
【化14】
【請求項11】
式(II)、(III)、(IV)、(V)、(VI)および(VIII)のいずれかの化合物の塩から選択される、請求項9に記載の化合物の塩:
【化15】
【請求項12】
式(XI)、式(XII)またはその塩から選択される化合物:
【化16】
【請求項1】
4−(3−クロロ−2−フルオロアニリノ)−7−メトキシ−6−{[1−(N−メチルカルバモイルメチル)ピペリジン−4−イル]オキシ}キナゾリンまたはその塩の製造方法であって:
(a)式(II)の化合物:
【化1】
を適切な酸の存在下で3−クロロ−2−フルオロアニリンと反応させること;または
(b)式(III)の化合物:
【化2】
を適切な酸の存在下で式(XI)または式(XII)の化合物:
【化3】
と反応させること;
を含む方法。
【請求項2】
酸が酢酸、ブタン二酸、フマル酸またはプロパン酸である、請求項1に記載の方法。
【請求項3】
式(II)の化合物が、式(III)の化合物:
【化4】
をN,N−ジメチルホルムアミドジメチルアセタールと反応させることを含む方法により製造される、請求項1に記載の方法。
【請求項4】
式(III)の化合物が、式(IV)の化合物:
【化5】
の還元を含む方法により製造される、請求項1または3に記載の方法。
【請求項5】
式(IV)の化合物が、式(V)の化合物:
【化6】
のニトロ化を含む方法により製造される、請求項4に記載の方法。
【請求項6】
式(V)の化合物が、式(VI)の化合物:
【化7】
を式(VII)の化合物:
【化8】
(式中、Lg1は適切な脱離基である)と反応させることを含む方法により製造される、請求項5に記載の方法。
【請求項7】
式(VI)の化合物が、式(VIII)の化合物:
【化9】
(式中、Pg1は適切なアミノ保護基である)の脱保護を含む方法により製造される、請求項6に記載の方法。
【請求項8】
式(VIII)の化合物が、式(X)の化合物:
【化10】
を式(IX)の化合物:
【化11】
(式中、Lg2は適切な脱離基であり、Pg1は適切なアミノ保護基である)と反応させることを含む方法により製造される、請求項7に記載の方法。
【請求項9】
式(XIII)の化合物またはその塩:
【化12】
(式中、R1は、H、−NH2、−NO2、または
【化13】
であり;
R2は、H、N−メチルカルバモイルメチル、またはPg1であり;
Pg1は、アミノ保護基であり;
ただし、R2がHまたはアミノ保護基である場合はR1はHである)。
【請求項10】
式(II)、(III)、(IV)、(V)、(VI)および(VIII)のいずれかの化合物から選択される、請求項9に記載の化合物:
【化14】
【請求項11】
式(II)、(III)、(IV)、(V)、(VI)および(VIII)のいずれかの化合物の塩から選択される、請求項9に記載の化合物の塩:
【化15】
【請求項12】
式(XI)、式(XII)またはその塩から選択される化合物:
【化16】
【図1】

【図2】
【図3】
【図4】

【図2】
【図3】
【図4】
【公表番号】特表2012−524769(P2012−524769A)
【公表日】平成24年10月18日(2012.10.18)
【国際特許分類】
【出願番号】特願2012−506579(P2012−506579)
【出願日】平成22年4月22日(2010.4.22)
【国際出願番号】PCT/GB2010/050653
【国際公開番号】WO2010/122340
【国際公開日】平成22年10月28日(2010.10.28)
【出願人】(300022641)アストラゼネカ アクチボラグ (581)
【Fターム(参考)】
【公表日】平成24年10月18日(2012.10.18)
【国際特許分類】
【出願日】平成22年4月22日(2010.4.22)
【国際出願番号】PCT/GB2010/050653
【国際公開番号】WO2010/122340
【国際公開日】平成22年10月28日(2010.10.28)
【出願人】(300022641)アストラゼネカ アクチボラグ (581)
【Fターム(参考)】
[ Back to top ]