
Fターム[4C057CC02]の内容
糖類化合物 (12,552) | Oがハロ、N、S、Se、Te、で置換された糖 (1,468) | ハロゲン (131)
Fターム[4C057CC02]に分類される特許
101 - 120 / 131
分子画像化プローブのクリックケミストリー合成法
本開示は、標的生体高分子に対し親和性を有する放射性リガンド又は放射性基質の調製方法であって、(a)クリックケミストリー反応に関与し得る第一の官能基を含んでなる第一の化合物と放射性試薬とを、脱離基を放射性試薬の放射性成分で置換するために十分な条件下、反応させて、第一の放射性化合物を形成すること;(b)第一の官能基とのクリックケミストリー反応に関与し得る第二の相補的官能基を含んでなる第二の化合物を提供すること;(c)第一の放射性化合物の第一官能基と第二の化合物の相補的官能基とをクリックケミストリー反応を介して反応させ、放射性リガンド又は基質を形成すること;及び(d)放射性リガンド又は基質を単離すること;を含んでなる方法を提供する。 (もっと読む)
抗HIV剤としてのヌクレオシドホスホネート結合体
本発明は、リン置換抗ウイルス阻害化合物、このような化合物を含有する組成物、および治療方法(これは、このような化合物を投与する工程を包含する)だけでなく、このような化合物を調製するのに有用なプロセスおよび中間体に関する。本発明は、HIV活性を備えた新規化合物、すなわち、新規ヒトレトロウイルスRTインヒビターを提供する。従って、本発明の化合物は、レトロウイルスRTを阻害し得、それゆえ、このウイルスの複製を阻害し得る。 (もっと読む)
2−デオキシ−L−リボフラノシルクロリド化合物の製造方法
【課題】 本発明は、工業的生産に適した、効率的な2−デオキシ−L−リボフラノシルクロリド化合物の製造方法、さらには該2−デオキシ−L−リボフラノシルクロリド化合物の製造に重要な中間体化合物および該中間体化合物の製造方法の提供。
【解決手段】 2−デオキシ−2−ハロ−L−アラビノフラノース化合物を脱ハロゲン化し、で表される2−デオキシ−L−リボフラノース化合物を得た後、該2−デオキシ−L−リボフラノース化合物を塩素化試薬と反応させることを特徴とする2−デオキシ−L−リボフラノシルクロリド化合物の製造方法。
(もっと読む)
オリゴヌクレオチド類似体
本発明は、一般的には、アプタマーを用いて免疫系を調節する方法に関し、とりわけ、CTLA−4機能を阻害する方法及びかかる方法に使用するのに適したアプタマーに関する。 (もっと読む)
ゲムシタビン誘導体の経口投薬形態
本発明は、2’,2’−ジフルオロデオキシシチジン(ゲムシタビン)のある特定の長鎖飽和脂肪酸誘導体及び長鎖モノ不飽和脂肪酸誘導体の経口投薬形態に関する。特に、本発明は、癌の治療コンプライアンスを改善させる経口投薬形態を製造するための上記ゲムシタビン誘導体又はそれらの医薬的に許容可能な塩の使用に関する。  (もっと読む)
(もっと読む)
βアノマーが富化された21−デオキシ−21,21−ジフルオロ−D−リボフラノシルヌクレオシドの調製のための中間体と方法
本発明は、βアノマーが富化された21−デオキシ−21,21−D−リボフラノシルジフルオロヌクレオシド〔式(II)〕、およびこれらの生理学的に許容できる塩、特に、ゲムシタビン塩酸塩〔式(IIb)〕のβ-富化されたアノマーを99%より高い純度で調製するための、式(I)の新規なトリクロロアセトイミダートを利用することによる、高選択的、簡単、かつ経済的なグリコシル化法を提供する。
 (もっと読む)
(もっと読む)
ヌクレオチド脂質エステル誘導体
本発明の対象は、特別なハロゲン化アデニンヌクレオチドの脂質エステル及びそのような脂質エステルの腫瘍治療における使用である。 (もっと読む)
2’−デオキシ−2’,2’−ジフルオロシチジンの製造方法
式(I)の2’−デオキシ−2’,2’−ジフルオロシチジンの製造方法を提供する。i)溶媒の中で式(III)の1−ハロリボフラノース化合物と式(IV)の核酸塩基を反応させて式(II)のヌクレオシドを得、反応中に生成される式(V)のハロゲン化シリルを連続的に除去する段階;及び
ii)式(II)のヌクレオシドを脱保護して式(I)の2’−デオキシ−2’,2’−ジフルオロシチジンを得る段階
を含む式(I)の2’−デオキシ−2’,2’−ジフルオロシチジンの立体選択的な製造。  (もっと読む)
(もっと読む)
アルコール溶媒中での有機フルオロ化合物の調製方法
本発明は、放射性同位元素フッ素18を含んだ有機フルオロ化合物の調製方法に関するもので、より詳細には、放射性同位元素フッ素18を含んだフッ素塩とアルキルハライドまたはアルキルスルホナートとを反応させて有機フルオロ化合物を調製する方法において、下記の化学式1で表わされるアルコールを溶媒に使用することによって、有機フルオロ化合物を高収率で調製する有機フルオロ化合物の調製方法に関するものであり、本発明による合成は、穏和な反応条件下で行うことができ、反応時間を短縮させるだけではなくその収率を顕著に向上させることにより、有機フルオロ化合物の大量生成に好適である。 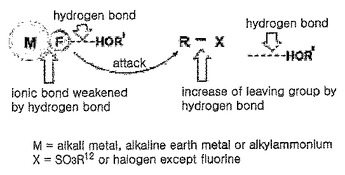 (もっと読む)
(もっと読む)
ジフルオロヌクレオシド及びその調製方法
2', 2'−ジフルオロヌクレオシドの立体選択的調製方法が提供される。この方法においては、保護された2', 2'−ジフルオロフラノースが、ルイス酸の存在下で、ピリミジン及びプリン誘導体から成る群から選択された塩基によりカップリングされ、ここで前記保護された2', 2'−ジフルオロフラノースは、1−位の脱離基及び3−及び5−位の保護基を有し、そして前記塩基が1又はそれ以上の酸素原子を含む場合、塩基は保護された塩基であり、個々の酢酸原子は保護基により保護されている。 (もっと読む)
フッ素化法

【解決手段】 非フッ素化糖誘導体とフッ化物との反応を含んでなるフッ素化糖誘導体の製造に際して、上記反応を1000ppm超50000ppm未満の量の水を含有する溶媒中で実施する。
【発明の効果】 本発明の方法では、制御された量の水の存在下では、反応収率は減少するどころか実際に増加する。反応混合物は1000ppmを超える量の水を含んでいなければならないので、反応混合物に存在する水の量を一定に保つのが格段に容易であり、反応条件を一貫して再現できる。従来技術の方法で用いられている乾燥工程の幾つかを省くことができ、試薬のコスト及び合成装置の製造コストに関してプロセス全体のコストが低減する。
(もっと読む)
N4−アセチル−2’−デオキシ−2’−フルオロ−シチジン誘導体の製造法
【課題】高純度なN4−アセチル−2’−デオキシ−2’−フルオロ−シチジン誘導体の製造法を提供すること
【解決手段】一般式(1)
(式中、R1は水素、炭素数1〜4のアルキル基、炭素数2〜4のアルケニル基、炭素数2〜4のアルキニル基、炭素数1〜4のペルフルオロアルキル基、またはハロゲン原子を表す。)で表される化合物に、包接用溶媒Qを含む溶媒中で該包接用溶媒を包接させる、一般式(2)
(式中、R1は前記の通りであり、m及びnはそれぞれ1〜100の整数であり、Qはエーテル系化合物、ケトン系化合物、またはエステル系化合物を表す。)で表される包接化合物の結晶の製造法。
(もっと読む)
脱保護の方法
本発明は、18F標識生成物の合成方法であって、弱酸を含む脱保護剤を使用して保護18F標識化合物を脱保護する段階を含み、脱保護生成物の中和及び緩衝化が中和剤の添加により行われる方法を提供するものである。脱保護生成物は、注射可能な放射性薬剤への続いてのオートクレーブ処理及び処方に適したpH範囲内に緩衝化される。
選択図【なし】
(もっと読む)
ApoBのRNAi調節およびその使用
本発明は、アポリポタンパク質Bの発現を調節するための組成物および方法、ならびにより詳細には化学的に修飾されたオリゴヌクレオチドによるアポリポタンパク質Bの下方制御に関する。 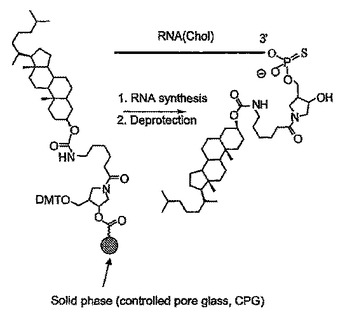 (もっと読む)
(もっと読む)
新しい芳香族フルオログリコシド誘導体、該化合物を含有する医薬品、およびその使用
本発明は式中の基が所定の定義の結合を有する式(I)の置換芳香族フルオログリコシド誘導体、生理学的に耐容性のあるその塩、および、その製造方法に関する。該化合物は例えば抗糖尿病剤の形態において使用できる。
【化1】
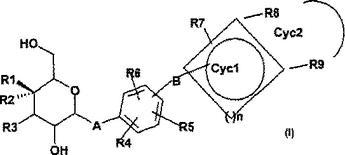 (もっと読む)
(もっと読む)
新しい複素環フルオログリコシド誘導体、該化合物を含有する医薬品、およびその使用
本発明は式中の基が所定の定義の結合を有する式(I)の置換フルオログリコシド複素環誘導体、生理学的に耐容性のあるその塩、および、その製造方法に関する。該化合物は例えば抗糖尿病剤の形態において使用できる。
【化1】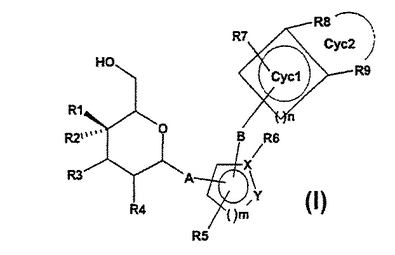
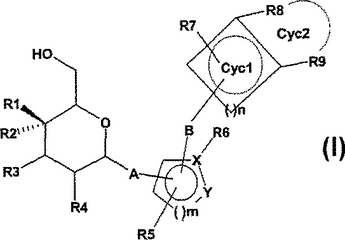 (もっと読む)
(もっと読む)
2’−フルオロ−2’−アルキル−置換又は他の置換されていてもよいリボフラノシルピリミジン類及びプリン類並びにそれらの誘導体の製造
本発明は、(i)2'-デオキシ-2'-フルオロ-2'-メチル-D-リボノラクトン誘導体、(ii)強い抗HCV活性を有するヌクレオシド類及びその類似体への中間体ラクトン類の転化、及び(iii)予め生成された好ましくは天然に生じるヌクレオシドから2'-デオキシ-2'-フルオロ-2'-C-メチル-β-D-リボフラノシルヌクレオシド類を含む抗HCVヌクレオシド類を製造する方法を提供する。 (もっと読む)
フッ素化プリンヌクレオシド誘導体の製造方法およびその中間体およびその製造方法
【課題】 工業的生産に適した、経済的かつ効率的な3’位(好ましくはα位)がフッ素化されたフッ素化プリンヌクレオシド誘導体の製造方法およびその中間体およびその製造方法を提供する。
【解決手段】 5’位が保護された新規な、8,2’−アンヒドロ−8−メルカプト−9−アラビノフラノシルプリン誘導体をフッ素化することによって、3’位がフッ素化された新規な8,2’−アンヒドロ−3’−デオキシ−3’−フルオロ−8−メルカプト−9−β−アラビノフラノシルプリン誘導体(2)が高収率で得られ、さらに、化合物(2)に対し、脱硫工程、R1の脱保護、又は必要に応じて核酸塩基の保護、脱保護、修飾を行うことにより、3’位がフッ素化された所望のプリンヌクレオシド誘導体を容易に合成することができる。
【化1】 (もっと読む)
(もっと読む)
アルキル置換された2−デオキシ−2−フルオロ−D−リボフラノシルピリミジン類及びプリン類及びそれらの誘導体の調製
本発明は、(i)2-デオキシ-2-フルオロ-2-メチル-D-リボノラクトン誘導体の製造方法、(ii)ラクトンの、強力な抗HCV活性を有するヌクレオシド、及びその類似体への転換、及び(iii)予め生成された、好ましくは自然に生じたヌクレオシドから2-デオキシ-2-フルオロ-2-C-メチル-β-D-リボフラノシルヌクレオシドを含む抗HCVヌクレオシドを製造する方法を提供する。 (もっと読む)
放射性材料のための可搬型製造設備
【課題】放射性医薬品を生成して包装する。
【解決手段】建造物がサイクロトロン(112)を収容すると共に、トラック又は列車で目的設置場所まで可搬性であるように設計され、製造設備(100)が、運搬中にサイクロトロンが無いことを除けば放射性医薬品を生成して包装するように実質的に装備されている製造設備を提供する。方法は、製造設備がサイクロトロンを受けるように設計する段階と、製造設備にサイクロトロンから第1の放射性材料を受け取り第2の放射性材料を生成する合成ユニット(132)を装備する段階と、製造設備を設置場所に運搬する段階と、サイクロトロンを設置場所に運搬する段階と、製造設備内にサイクロトロンを密閉する段階とを含む。製造設備は、あまり労力を用いること無く、別の設置場所に再配置することができる。 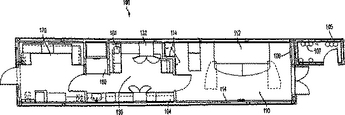 (もっと読む)
(もっと読む)
101 - 120 / 131
[ Back to top ]