
皮膚疾患の予防または治療用のバルプロ酸の局所使用
本発明は、単独またはレチノイドもしくは核内受容体リガンド、または化学療法剤(例えば、5−フルオロウラシル)の局所適用製剤と組合せて使用されうるバルプロ酸またはその誘導体を含有する局所適用製剤に関する。本製剤は、基底細胞癌、扁平上皮細胞癌、角化棘細胞腫、ボーエン病、皮膚T細胞リンパ腫など癌性皮膚疾患の局所治療とともに、皮膚および/または粘膜の前悪性病変、および炎症の局所治療に有用である。本発明は、紫外線からの保護用、および日焼けの治療用の本局所適用製剤の使用にも関する。本発明は、上記のヒト疾患の局所治療用に臨床的に使用される医薬製造用のVPAの使用を含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ヒト癌、例えば基底細胞癌、扁平上皮細胞癌、角化棘細胞腫、ボーエン病、皮膚T細胞リンパ腫などの局所治療用とともに、前悪性病変(光線性角化症など)、および皮膚および/または粘膜の炎症(乾癬、魚鱗癬、座瘡など)の局所治療用の単独またはレチノイドもしくは核内受容体リガンド、または化学療法剤(例えば、5−フルオロウラシル)の局所適用製剤と組合せて使用されうるバルプロ酸またはその誘導体を含有する局所適用製剤に関する。本発明は、紫外線からの保護用、および日焼けの治療用の本局所適用製剤の使用にも関する。また、本製剤は、ヒストンの低アセチル化と関係があり、または高アセチル化の誘導が、例えば、形質転換皮膚および粘膜細胞における分化および/またはアポトーシスの誘導によって有利な効果を有する疾患において使用されうる。さらに、本製剤は、皮膚および/または粘膜の他の増殖性疾患の治療、および皮膚および/または粘膜組織における異常な遺伝子発現と関係がある状態の治療または予防に有用である。本発明は、上記のヒト疾患の局所治療用に臨床的に使用される薬品の製造を含む。
【背景技術】
【0002】
クロマチン制御と癌
クロマチンの局所再構成は、遺伝子の転写活性化における重要なステップである。DNAのヌクレオソームパッケージングにおける動的変化が生じ、転写タンパク質をDNA鋳型と接触させる必要がある。クロマチン再構成および遺伝子転写に影響を及ぼす最も重要な機序の1つは、クロマチン構造におけるアセチル化およびその後の変化によるヒストンおよび他の細胞タンパク質の翻訳後修飾である(デービー(Davie)、1998年、Curr Opin Genet Dev 8、p.173−178、コウザライズ(Kouzarides)、1999年、Curr Opin Genet Dev 9、p.40−48、ストラール(Strahl)とアリス(Allis)、2000年、Nature 403、p.41−44)。ヒストン高アセチル化の場合、DNAに対する静電引力の変化および疎水性アセチル基によって導入される立体障害が、DNAとのヒストンの相互作用の不安定化をもたらす。結果として、ヒストンのアセチル化がヌクレオソームを崩壊させ、DNAが転写機構にアクセスしやすくなることを可能にする。アセチル基の除去は、ヒストンがDNAおよび隣接したヌクレオソームとよりしっかりと結合し、したがって転写抑制されたクロマチン構造を維持することを可能にする。アセチル化は、ヒストンアセチルトランスフェラーゼ(HAT)活性を有する一連の酵素によって介在される。逆に言えば、アセチル基が特異的ヒストンデアセチラーゼ(HDAC)酵素によって除去される。これらの機序の崩壊は転写ミスレギュレーションを生じさせ、腫瘍化および腫瘍進行の原因となりうる。
【0003】
また、転写因子など他の因子は、そのアセチル化状態によって活性および安定性を変化させる。例えば、急性前骨髄球白血病(APL)と関係がある融合タンパク質であるPML−RARは、p53の脱アセチル化および分解を介在することによりp53を阻害し、したがってAPLブラストがp53依存癌監視経路を回避することを可能にする。造血前駆細胞におけるPML−RARの発現は結果として、p53介在転写活性化の抑制、および遺伝毒性ストレス(X線、酸化的ストレス)によって誘因されるp53依存アポトーシスからの保護をもたらす。しかし、p53の機能は、p53阻害の基礎を成す機序としてPML−RARによるp53へのHDACの積極的関与にかかわるHDAC阻害剤の存在下に再インストールされる(インシンガ(Insinga)ら、2003年、投稿中)。したがって、タンパク質のアセチル化は、p53のアセチル化などヒストンと異なり、HDAC阻害剤の抗腫瘍活性における重要な役割を果たす。
【0004】
核内受容体とヒストンデアセチラーゼ
核内ホルモン受容体は、遺伝子発現の陽性および陰性対照により発生および恒常性を制御するリガンド依存転写因子である。これらの制御過程における欠陥が、多くの疾患の原因の基礎を成し、癌の発生における重要な役割を果たす。T3R、RAR、およびPPARを含む多くの核内受容体は、リガンドの非存在下にN−CoRおよびSMRTなどコリプレッサーと相互作用し、それによって転写を阻害しうる。さらに、N−CoRは、アンタゴニストと結合したプロゲステロンおよびエストロゲン受容体と相互作用することも報告されている。最も興味深いことに、N−CoRおよびSMRTは、mSin3タンパク質およびヒストンデアセチラーゼをも含有する大きなタンパク質複合体に存在することが示されている(パジン(Pazin)とカドナガ(Kadonaga)、1997年、Cell 89、p.325−328)。したがって、核内受容体の抑制から活性化へのリガンド誘発スイッチは、コリプレッサーおよび補助活性化因子の複合体の拮抗酵素活性への変換を示す。
【0005】
核内受容体による遺伝子制御
HDAC活性を含有するかかるコリプレッサー複合体は、核内受容体による抑制を介在するだけではなく、Mad−1、BCL−6、およびETOを含む別の転写因子と相互作用もする。これらのタンパク質の多くは、細胞増殖および分化の疾患において重要な役割を果たす(パジン(Pazin)とカドナガ(Kadonaga)、1997年、Cell 89、p.325−328、ヒューン(Huynh)とバードウェル(Bardwell)、1998年、Oncogene 17、p.2473−2484、ワング(Wang),J.ら、1998年、Proc Natl Acad Sci USA 95、p.10860−10865)。例えば、T3Rは最初は、野生型受容体とは対照的にリガンドに結合することはなく、転写の構成的コリプレッサーとして機能するそのウイルス癌遺伝子v−erbAとの相同性に基づき同定された。さらに、RARにおける突然変異は、多くのヒト癌、特に急性前骨髄球性白血病(APL)および肝細胞癌と関係がある。APL患者においては、染色体転座由来のRAR融合タンパク質が、前骨髄球白血病タンパク質(PML)または前骨髄球亜鉛フィンガータンパク質(PLZF)のいずれかを伴う。両方の融合タンパク質はコリプレッサー複合体の成分と相互作用しうるが、レチノイン酸の添加によりPML−RARからコリプレッサー複合体が追放されるの対して、PLZF−RARは構成的に相互作用する。これらの所見は、PML−RARAPL患者がレチノイン酸治療後に完全寛解を達成するのに対して、PLZF−RAR APL患者がきわめて不十分に反応する理由の説明を提供する(グリニャーニ(Grignani)ら、1998年、Nature 391、p.815−818、グイデ(Guidez)ら、1998年、Blood 91、p.2634−2642、ホー(He)ら、1998年、Nat Genet 18、p.126−135、リン(Lin)ら、1998年、Nature 391、p.811−814)。
【0006】
最近、レチノイン酸による治療後に多重再発を経験したPML−RAR患者が最近、HDAC阻害剤のフェニルブチレートで治療され、結果として白血病の完全寛解がもたらされている(ワレル(Warrell)ら、1998年、J.Natl.Cancer Inst.90、p.1621−1625)。
【0007】
HDAC阻害剤による抗癌治療
追加の臨床試験では最近、HDAC阻害の原理を用いた癌患者の全身臨床治療の活用が開始されている。今のところ、単剤療法としての密接に関連した酪酸誘導体ピバネックス(Pivanex)(チタン製薬(Titan Pharmaceutical))による第II相臨床試験が完了し、第III/IV期の非小細胞肺癌における活性が明らかにされている(キール(keer)ら、2002年、ASCO、要約番号1253)。多くのHDAC阻害剤が同定されており、NVP−LAQ824(ノバルティス(Novartis)およびSAHA(アトンファルマ(Aton Pharma)社)が第II相臨床試験されたヒドロキサム酸の構造的クラスのメンバーである(マークス(Marks)ら、2001年、Nature Reviews Cancer 1、p.194−202)。他のクラスは、T細胞リンパ腫の治療用に第II相臨床試験で有効に使用されたデプシペプチド(FR901228号−藤沢(Fujisawa))など環状テトラペプチドを含む(ピエカルツ(Piekarz)ら、2001年、Blood 98、p.2865−2868)。さらに、ベンズアミドのクラスに関連した化合物であるMS−27−275(三井製薬(Mitsui Pharmaceuticals))が現在、悪性血液疾患患者を治療する第I相治験で試験されている。
【0008】
ヒストンデアセチラーゼのタンパク質ファミリー
哺乳動物のヒストンデアセチラーゼは3つのサブクラスに分類されうる(グレイ(Gray)とエクスレーム(Ekstroem)、2001年)。酵母RPD3タンパク質のホモローグであるHDAC1、2、3、および8は、クラスIを構成する。HDAC4、5、6、7、9、および10は酵母Hda1タンパク質と関係があり、クラスIIを形成する。最近、NAD依存であるデアセチラーゼの第IIIクラスを形成する酵母Sir2タンパク質の一部の哺乳動物のホモローグが同定されている。これらHDACのすべては、多くのマルチタンパク質複合体のサブユニットとして細胞に存在すると思われる。具体的には、クラスIおよびIIにHDACは、転写因子へのHDACの動員に必要な架橋因子として使用される転写補体mSin3、N−CoR、およびSMRTと相互作用することが示されている。
【0009】
バルプロ酸
バルプロ酸(VPA、2−プロピル−ペンタン酸)は、異なる分子作用機序に基づく多くの生物活性を有する。すなわち、
−VPAは、抗てんかん薬である。
−VPAは、催奇性である。妊娠中の抗てんかん薬として使用される場合、VPAは生誕児の数パーセントにおいて出生異常(神経管閉鎖障害および他の奇形)を誘発しうる。マウスにおいては、VPAは、適切に投与された場合に大半のマウス胎児において催奇性である。
−VPAは、核内ホルモン受容体(PPARδ)を活性化するいくつかの追加の転写因子はまた抑制されるが、ある因子は顕著には抑制されない(グルココルチコイド受容体、PPARα)。
−VPAは、場合により、補酵素Aにより十分に代謝されないエステルに依存した肝毒性を引き起こす。
−VPAは、HDACの阻害剤である。
【0010】
VPA誘導体の使用により、異なる活性が異なる分子作用機序によって介在されることが判明した。催奇性および抗てんかん活性は、選択的に催奇性または選択的に抗てんかんである化合物が単離されうるため、異なる作用形態をたどる(ナウ(Nau)ら、1991年、Pharmacol.Toxicol.69、p.310−321)。PPARδの活性化は、催奇性と厳密に相関し(ランペン(Lampen)ら、1999年、Toxicol.Appl.Pharmacol.、160、p.238−249)、PPARδの活性化および催奇性の両方がVPAの同じ分子活性を必要とすることが示唆された。また、F9細胞の分化は、ランペン(Lampen)ら、1999年によって示差され、かつ分化マーカーの分析によって立証されているように、PPARδの活性化および催奇性と厳密に相関する(ウェルリング(Werling)ら、2001年、Mol.Pharmacol.59、p.1269−1276)。PPARδの活性化は、VPAおよびその誘導体のHDAC阻害活性によって引き起こされることが示された(PCT/EP01/07704号、国際公開第03/024442A2号パンフレット)。さらに、既定のHDAC阻害剤であるTSAがPPARδを活性化し、かつVPAと同じ型のF9細胞分化を誘発することが示された。これらの結果から、PPARδの活性化だけではなく、F9細胞分化の誘発およびVPAまたはVPA誘導体の催奇性もHDAC阻害によって引き起こされると結論できる。
【0011】
抗てんかんおよび鎮静活性は、異なる構造活性相関をたどり、したがって、明らかにHDAC阻害と異なった一次VPA活性に依存する。肝毒性の機序の理解は不十分であり、VPA−CoAエステルの形成と関係があるかどうかは不明である。しかし、HDAC阻害は、CoAエステル形成を必要とするようには思われない。
【0012】
レチノイン酸
レチノイン酸(RA)は、遺伝子転写の調整において重要な役割を果たし、したがって、細胞分裂および分化、免疫応答および胚発生など体内の多数の機能を支配する、9−cisRA、トランスRA、全トランスRA、およびタザラトン(Tazaraton)を含むがこれらに限定されないレチノイドとして知られる化合物のクラスに属する(ギュンター(Guenther)、2003年、Am.J.Clin.Dermatol.4、p.197−202、ペリス(Peris)Kら、1999年、N Engl J Med 341、p.1767−1768)。それらはまた、癌細胞の発生および拡大を制御し、RAを含む一部は、癌細胞増殖を阻止することによって腫瘍増殖を阻害する(アルトゥッチ(Altucci)とグロンマイヤー(Gronemeyer)、2001年、Nature Reviews Cancer 1、p.181−193)。
【0013】
レチノイドの効果は2つのクラスの核内受容体、RA受容体(RAR)およびレチノイドX受容体(RXR)によって主として介在される(チャン(Zhang)とファール(Pfahl)1993年、ケストナー(Kestner)ら、1995年、マンゲルスドルフ(Mangelsdorf)とエバンス(Evans)1995年)。RARおよびRXRは、3つの異なる遺伝子(α、β、およびγ)によって暗号化される。また、多くのレチノイド受容体イソ型は、分化プロモーターの使用により生成され、多数の異なるレチノイド受容体タンパク質を生じさせる。今日まで、レチノイドの効果を介在することが知られている多数の受容体がある。9−cisRAは、RARおよびRXRの高アフィニティーリガンドであるが、トランス−RAはRARのみのリガンドである。レチノイド受容体は、多くのホルモン、ビタミン、および薬剤の生物学的効果を介在する大きなステロイド/甲状腺受容体スーパーファミリーに属する。RARおよびRXRは転写因子として作用し、活性クロマチン構造の標的遺伝子のプロモーター領域に位置したその応答エレメント(RARE)に結合することによって標的遺伝子の発現を積極的または消極的に制御し、結果として標的遺伝子の転写をもたらす。転写に対するその直接効果に加えて、リガンド化RARは、AP−1など他の転写因子の活性を調節しうる(ファール(Pfahl)1993年)。活性化レチノイド受容体はAP−1の活性を阻害し、それによってAP−1標的遺伝子の発現を制御しうる。AP−1活性の阻害は、レチノイドの抗増殖性効果と関係があり、レチノイド標的遺伝子の転写のその直接の活性化とは別であるように思われる。
【0014】
かねて医師はRA誘導体を用いていくつかの癌、特に前立腺癌および白血病を治療しており、現在ではこの薬剤で乳癌を治療する実験中である。RAによる典型的な治療では好ましい遺伝子に切り替えるためにRARを活性化することが追求される。しかし、RAに対する大きな欠点は、遺伝子を「オン」および「オフ」にするために高レベルの薬物投与を必要とすることであり、これはしばしば破壊的かつ潜在的に致死的な副作用を誘因する(アルトゥッチ(Altucci)とグロンマイヤー(Gronemeyer)、2001年、Nature Reviews Cancer 1、p.181−193)。
【0015】
RAまたはタザロテンなど他のレチノイド(ギュンター(Guenther)、2003年、Am.J.Clin.Dermatol.4、p.197−202)も軽度〜中程度の座瘡および日焼けによる損傷(光老化)皮膚の治療に局所使用されうる。レチノイドは、基底細胞癌など一部の皮膚癌を治療するために使用されている(ペリス(Peris)Kら、1999年、N Engl J Med 341、p.1767−1768)。局所投与後、RAは細胞分裂および代謝回転を増大させるように思われる。皮膚の外側部分における細胞層の数は減少する。吹出物が存在する場合、それらはより迅速に消散する。局所RAは、細いしわ、シミだらけの高色素沈着、および日光への露出過度と関係した粗さの削減において有効である。治療の結果は即効性はなく、数週間〜数か月後に明らかとなる。治療の中断は通常、結果的にRA効果の喪失をもたらす。
【0016】
基底細胞癌
基底細胞癌(BCC)は皮膚癌のもっとも一般的な形態であるだけではなく、例えば、毎年80万のアメリカ人に影響を与えるすべての癌のうち最も一般的でもある。これらの癌は、表皮(外側皮膚層)の一番下にある基底細胞に生じる。新しい症例の数は過去数十年間に毎年急に増大したが、疾患の平均発症年齢は確実に低下した。最近まで、最も多く罹患するのは高齢者、特に屋外労働していた男性であった。しかし、今日では多くの女性が過去におけるよりもBCCに罹っているが、それでも男性が依然として女性の数を大きく上回っている。
【0017】
日光への慢性的な被曝は、体の露出部分―顔、耳、首、頭皮、肩、および背中で最も頻繁に生じるほぼすべての基底細胞癌の原因である。白い肌、色の薄い髪、青色、緑色、または灰色の眼は、きわめてリスクが高い。職業が長時間屋外にいることを必要とする、または日なたで多大な余暇時間を過ごすような人は特に危険である。BCCの素因としては、紫外線、電離放射線、ヒ素、色素乾皮症および基底細胞母斑症候群などの遺伝性疾患、ワクチン接種、火傷、および瘢痕が挙げられる。
【0018】
典型的な病変は、隆起した、真珠状または半透明の境界を有する滑面結節である。病変における毛細血管拡張症が一般的な特徴である。基底細胞癌はサイズが数ミリメートル〜数センチメートルのばらつきがある。サイズは6−12か月で2倍になりうるし、または増殖がゆっくりでありうる。85パーセントが頭および首に生じ、鼻は最も一般的な部位である。患者は、手ぬぐいでの洗浄など小さな外傷による出血を報告する場合もある。この新生物は局所侵襲的であり、眼、耳、または鼻の喪失を引き起こすきわめて破壊的であり、かつ脳に侵入する場合には致死的でありうる。BCCはまれにしか再発しないが、再発する場合、リンパ管を経由して局所リンパ節に、または血行性散布によって最も一般的には肺に転移しうる。進行した皮膚癌は任意に2cmより大きい腫瘍とともに、骨筋、もしくは神経の侵襲、リンパ節転移、または美容または機能単位の除去を必要とする病変として定義されうる。
【0019】
BCCの治療は、その型、サイズ、位置、治療される数、および医師の選好または経験によって決まる。可能性としては、以下が挙げられる。すなわち、
・切除術―病変は切除され、皮膚は縫合される。
・凍結処理(凍結療法)―小さな表在性病変用に液体窒素の使用。これはBCC用の推奨治療ではない。
・掻爬術および焼灼―腫瘍の掻爬および基底の焼灼。これもBBC用の推奨治療ではない。
・放射線療法(X線治療)―進行した病変において、または手術が適切でない場合に使用されうる。
・局所化学療法―5−フルオロウラシル(5−FU)含有クリームの局所適用は、特に手術によって治療可能ではないきわめて大きな部位用に使用されうる。
【0020】
しかし、手術を嫌う、または手術が不適切な患者(例えば、ペースメーカーを付け、または出血性素因を有する虚弱で高齢の患者)、または大きな病変、多数の病変、および解剖学的に困難な部位または不十分な血管化新生皮膚を有する患者については、依然として別の有効な治療法を必要とする。
【0021】
今日、2つ以上の抗腫瘍治療用薬剤による患者の組合せ治療から成る腫瘍治療が知られている。例は、化学療法剤および/または細胞毒性試薬とともに放射線療法の併用、および腫瘍細胞特異的治療抗体の使用など、免疫学的療法と放射線療法のより最近の組合せである。しかし、個々の方法の単独よりも有効であるかかる組合せを確認するための個々の治療を互いに組合せる可能性は、広範な前臨床試験および臨床試験を必要とする。どの組合せが付加的またはさらに相乗的効果を示すか予測することは可能ではない。治療効果を増大させる目的に加えて、他の目的が、個々の成分の高用量によって引き起こされる望ましくない、または有害な副作用を減少させるために結果として生じる組合せにおける個々の成分の用量の可能な減少である。
【0022】
自己免疫および炎症におけるサイトカインの役割
サイトカインは、個々の細胞および組織の機能的活性を調節する調整剤として作用する可溶性タンパク質およびペプチドの多様なグループである。それらは、外来物質または変化した内因性物質に対する防御として炎症反応を誘発させることを目的とするものである。多くの点で、サイトカインの生物活性は、炎症、急性期反応、および自己免疫など生物学的現象を誘発する全身レベルで作用することによって特殊な腺組織における生じる典型的なホルモンのものに似ている。しかし、炎症反応の不適切な活性化は多くの一般的な疾患の基礎を成す原因であり、したがって、炎症反応は薬剤開発の重要な目標である。
【0023】
多くのサイトカインが炎症を加速させ、直接または一部の細胞型における細胞接着分子または他のサイトカインの合成を誘発するその能力により局所または全身の炎症反応を調節する。初期反応の原因となる主なサイトカインは、IL1、IL6、およびTNF−アルファである。他の炎症誘発媒体としては、LIF、IFN−ガンマ、GM−CSF、IL11、IL12、IL18、およびさまざまな他のケモカインが挙げられる。
【0024】
しかし、サイトカインの役割は、炎症過程のみに限定されるのではなく、自己免疫疾患の発症および伝播において主要な役割も有する。古典的な例が関節リウマチであり、この場合、特異的CD4+T細胞が、最も可能性があるのは未知の外生または内因性抗原に対する反応として、罹患関節において免疫応答を誘発する(オルセン(Olsen)ら、2003年、New England Journal of Medicine 350、p.2167−2179)。したがって、増強された単球、マクロファージ、および線維芽細胞は、滑液腔内の腫瘍壊死因子−α(TNF−α)およびインターロイキン−1などのサイトカインを生じる。これらのサイトカインはカスケード損傷の中核を成し、最終的にマトリクスメタロプロテイナーゼおよび破骨細胞の産生を誘因し、これが結果として軟組織および骨の不可逆性損傷をもたらす。
【0025】
活性化単球、マクロファージ、およびTリンパ球によって放出される炎症性サイトカインであるTNF−αは、関節リウマチの病因において重要である炎症反応を促進する。関節リウマチ患者は滑液中に高濃度のTNF−αを有する。TNF−αは、炎症性パンヌスおよび健康な軟骨の接合部に局在されており、高い滑液TNF−α濃度は骨の侵食と関係がある。
【0026】
当然のことながら、TNF拮抗薬は、関節リウマチ用に利用可能な特に最も有効な治療薬であるように思われる。反応は一般に迅速であり、しばしば数週間以内に発生するが、すべての患者が反応を示すわけではない。
【0027】
TNF−αに対する薬剤は、関節リウマチなど慢性自己免疫疾患の治療において有効であるだけではなく、クローン病、潰瘍性大腸炎、シェーグレン症候群、強皮症、乾癬性関節炎、強直性脊椎炎、難治性ブドウ膜炎、ベーチェット病、成人発症型スティル病、およびヴェーゲナー肉芽腫症の治療においても有効である。
【0028】
別の例が乾癬であり、この場合、T細胞介在免疫応答がケラチン生成細胞に対して行われる。これらのTリンパ球は真皮または表皮において開始抗原に直面し、1型サイトカイン(Th1)、特にインターフェロン−γ、インターロイキン2、およびTNF−αを分泌する。これらの分泌が結果としてケラチン生成細胞の増殖および成熟低下とともに関連血管変化をもたらす。インターロイキン8など他のサイトカインの分泌は、乾癬の全体像の一因となる(レブボール(Lebwohl)、2004年、The Lancet 361、p.1197−1204)。
【0029】
自己免疫疾患におけるサイトカインの因果関係の別の証拠は、さまざまな疾患の治療におけるサイトカインの使用後に得られた所見に由来する(クラウゼ(Krause)ら、2003年、The American Journal of Medicine 115、p.390−397)。興味深いことに、それらは免疫および自己免疫状態の誘因および悪化などの副作用と関係があり、これらは明白な自己免疫疾患に発展しうる。これらの自己免疫症状は、自己免疫に対する既存の傾向を有する患者においてより一般的であるように思われる。
【0030】
多発性硬化症の悪化がインターフェロン−γによる治療中に観察されている。インターフェロン−γ治療と関係した自己免疫症状の頻度は低いように思われるが、骨髄増殖性疾患用にインターフェロン−γ単独、およびインターフェロン−γと組合せて治療された患者における全身紅斑性狼瘡が報告されている。インターフェロン−γは動物モデルにおける全身紅斑性狼瘡の病因に関与している。インターフェロン−γの投与は、抗インターフェロン−γ抗体による治療によって予防される狼瘡傾向のある(NZBXNZW)F1マウスにおける糸球体腎炎への進行速度を加速する。インターフェロン−γの血清レベルの上昇が、全身紅斑性狼瘡患者において報告されている。インターフェロン−γはナチュラルキラー細胞によって生成され、II型インターフェロン受容体に結合する。これはナチュラルキラー細胞の活性化においてインターフェロン−γよりも有効ではなく、強力な抗ウイルスおよび抗腫瘍効果を有さない。しかし、インターフェロン−γは、マクロファージ活性化および腫瘍組織適合性クラスII分子の最も強力な誘発因子である。これはB細胞による免疫グロブリン分泌を刺激し、Tヘルパー1型へのT細胞の分化を促進する。
【0031】
インターロイキン2は、抗腫瘍活性を有する活性化T細胞によって分泌される。これは転移性悪性黒色腫および腎細胞癌治療において有効である。これはT細胞増殖を誘発し、B細胞増殖を増強し、ナチュラルキラー細胞および単球の活性化を促進する。インターロイキン2治療下に確認される最も一般的な自己免疫副作用は、免疫介在甲状腺疾患である。可逆性甲状腺機能不全は、インターロイキン単独またはリンホカイン活性化キラー細胞もしくはインターフェロン−γとともにインターロイキンで治療される癌患者において頻繁に生じる。転移性腎細胞癌患者におけるインターロイキン2による試験では、患者の18%(60/329)において抗甲状腺抗体が検出された。自己免疫とみなされうる他のそれほど多くない現象が、インターロイキン2治療と関係して報告されている。これらには関節リウマチ、乾癬性関節炎、強直性脊髄炎、およびライター症候群が含まれる。関節炎の誘因は、関節を浸潤し、炎症をもたらすT細胞によって認識される自己抗原の誘導によって説明されうる。インターロイキン2は、筋特異的および腫瘍抗原に対する免疫寛容の崩壊を強化し、腫瘍および筋細胞の破壊をもたらす。インターロイキン2およびリンホカイン活性化キラー細胞で治療された転移性腎細胞癌の患者が全身性硬化の急性悪化を生じた。
【0032】
インターロイキン2および可溶性インターロイキン2受容体の血清レベルは、全身性硬化症患者において上昇し、疾患期間および活性と相関する。これらの所見によりインターロイキン2治療と全身性硬化症の発症との関係が説明されうる。
【発明の開示】
【発明が解決しようとする課題】
【0033】
本発明の目的は、皮膚疾患の予防または治療用の改善された組成物を提供することである。
【課題を解決するための手段】
【0034】
VPAは、てんかんの治療に使用される薬品として開発されている。したがって、VPAは、経口または静脈内に全身使用され、薬品が血液脳関門を通り脳組織のてんかん標的部位に達し、その抗てんかん使命を果たすことを可能にする。それゆえ、VPAは、その治療効果を達成するために全身適用されるべき薬品とみなされている。
【0035】
現在、皮膚の局所に使用される治療化合物に決定的に必要とされるVPAがヒト皮膚に有効に透過することが意外にも確認された。したがって、VPAは、局所適用のこの基本的な基準を満たすことによって、皮膚の癌病変の局所治療に使用されうることが本発明者らによって仮説として提出された。
【0036】
この結果、現在、VPAは実際に、ヒト皮膚疾患、例えば、ヒト皮膚癌の局所治療に使用されうると予期しない有利な効果を有することがわかった。ここで、VPAによって使用される正確な作用形態は完全には理解されていないが、アポトーシス、成長停止、および分化誘発化合物の活性への腫瘍細胞を感作するその可能性は、かかる抗腫瘍活性の基礎でありうる。VPAのこの意外な可能性は、HDAC活性を有する一連の特異的酵素の阻害剤としてのその活性に基づくと予想される。
【0037】
したがって、本発明は、皮膚疾患の予防または治療用の局所医薬組成物に関し、
(i)VPAおよびその医薬的に許容される塩、VPAの誘導体およびその医薬的に許容される塩から成る群より選択される活性剤の少なくとも約0.1%と、
(ii)皮膚科学的に許容される担体と
を含む。
【発明を実施するための最良の形態】
【0038】
本明細書で使用される「局所適用」という語は、本発明の組成物を皮膚の表面上に適用または塗布することを意味する。
【0039】
本明細書で使用される「皮膚科学的に許容される」という語は、そのように記載される組成物またはその成分が、過度の毒性、不適合性、不安定性、アレルギー反応なしにヒト皮膚と接触して使用するために適切であることを意味する。
【0040】
別段の指示がない限り、本明細書で示されているパーセント値は重量%(w/w)である。
【0041】
本発明の組成物は、好ましくは、活性剤の約0.1%〜約25%、より好ましくは、約0.1%〜約6%、さらにより好ましくは、約0.3%〜約5%、さらにより好ましくは、約0.5%〜約4%、さらにより好ましくは、約1%〜約4%、最も好ましくは、約2%〜約4%の活性剤を含む。
【0042】
本発明の組成物は通常、活性剤、および他の任意の活性物が適切な濃度で皮膚に送達されることを可能にする、本発明の組成物が組込まれている皮膚科学的に許容される担体の約1%〜約99.9%を含む。
【0043】
好ましい実施態様においては、本発明の組成物は、25℃および大気圧で半固体である。本実施態様によれば、組成物の製品形態は、クリーム、軟膏、ゲル、またはペーストであってよい。組成物の製品形態は液分散、例えば、ローションである。
【0044】
意外にも、VPAの局所適用は、レチノイド、および5−フルオロウラシルなどの化学療法剤の治療効果を増強することがわかっている。
【0045】
本発明の他の態様においては、したがって、レチノイン酸またはその誘導体をさらに含む組成物中のレチノイン酸または誘導体の濃度は、好ましくは、組成物の約0.01%〜約1%、より好ましくは、約0.05%〜約0.5%である。レチノイドは、好ましくは、9−シスレチノイン酸、トランス−レチノイン酸、全トランスレチノイン酸、およびタザロテンから成る群より選択される。
【0046】
本発明のさらに他の態様においては、組成物は、5−フルオロウラシルなどの化学療法剤をさらに含む。化学療法剤の濃度は、好ましくは、組成物の約0.1%〜約10%、より好ましくは、約1%〜約10%である。
【0047】
好ましい実施態様においては、担体は溶液ではない。他の好ましい実施態様においては、担体はクリーム、ペースト、軟膏、ローション、またはゲルである。
【0048】
本発明の他の態様は、少なくとも約0.1%のVPAまたはその誘導体を含んでなる医薬品がそれを必要とする個人の皮膚に局所適用される、皮膚疾患の予防または治療のための医薬品を製造するためのVPA、その医薬的に許容される塩、VPAの誘導体、またはその医薬的に許容される塩の使用である。
【0049】
皮膚疾患は、好ましくは、タンパク質の高アセチル化の誘導が患者に対する有利な治療効果を有するヒト皮膚の疾患である。皮膚疾患は、皮膚腫瘍、例えば、基底細胞癌、扁平上皮細胞癌、角化棘細胞腫、ボーエン病、および皮膚T細胞リンパ腫である。
【0050】
皮膚疾患は、光線性角化症などの前腫瘍性皮膚疾患でありうる。他の実施態様においては、VPAまたはその誘導体は、皮膚および/または粘膜の炎症の治療に使用されうる。皮膚および/または粘膜の炎症の限定されない例としては、乾癬、魚鱗癬、および座瘡である。皮膚疾患は太陽で痛んだ皮膚(光老化皮膚、日焼け)でもありうる。
【0051】
本発明によるVPAまたはその誘導体の投与は、確立された抗癌治療と組合せられうる。VPAまたはその誘導体および確立された癌治療は、同時に、または連続的に(異なる時点で)適用されうる。
【0052】
別の任意の活性物質が本発明による治療において使用されうる。VPAまたはその誘導体および別の活性物質を、同時に、または連続的に(異なる時点で)投与することができる。別の活性物質としては、NVP−LAQ824(ノバルティス(Novartis))、トリコスタチンA、スベロイルアニリドヒドロキサム酸(アトン(Aton))、CBHA (アトン(ATON))、ピロキサミド(Pyroxamide)(アトン(Aton))、スクリプタイド(Scriptaid)(ジョーンズ・ホプキンス(Johns Hopkins))、CI−994 (ファイザー(Pfizer))、CG−1521 (キルカゲン(CircaGen))、クラミドシン(Chlamydocin)(ヤンセン(Janssen))、ビアリールヒドロキサム酸塩、例えば、A−161906 (アボット(Abbott))、二環式アリール−N−ヒドロキシカルボキサミド(関西大学(Kansai University))、PXD−101(プロリフィックス(Prolifix))、スルホンアミドヒドロキサム酸(メチルジーン(MethylGene))、TPX−HA類似体(CHAP)(ジャパンエナジー(Japan Energy))、オキサムフラチン(Oxamflatin)、トラポキシン(Trapoxin)、デプデシン(Depudecin)、アピジシン(Apidicin)(キョンジ(Kyongji))、ベンズアミド、例えばMS−27−275(三井(Mitsui))、ピロキサミド(Pyroxamide)およびその誘導体、短鎖脂肪酸、例えば酪酸、およびその誘導体、例えば、ピバネックス(Pivanex)(ピバロイルオキシメチルブチレート)、環状テトラペプチド、例えばトラポキシンA、デプシペプチド(FK−228、藤沢(Fujisawa)/NCI)および関連ペプチド化合物、タセジナリン(Tacedinaline)(ファイザー(Pfizer))、MG2856(メチルジーン(MethylGene))、およびHDACクラスIII阻害剤またはSIRT阻害剤(ケリー(Kelly)、オコナー(O’Connor)とマークス(Marks)、2002年、Expert Opin.Inverstig.Drugs 11(12)、p.1695−1713を参照)などの化合物を含むがこれらに限定されないVPAとは異なるヒストンデアチラーゼの阻害剤が挙げられる。
【0053】
VPAまたはその誘導体の投与は、化学療法剤または細胞毒性剤(例えば、5−FU)、分化誘導剤(例えば、ビタミンD、ビタミンD誘導体、レチノイド、イミキモド(imiquimode)などの受容体結合剤)、放射線治療(例えば、エックス線またはガンマ線)、免疫学的方法(抗体治療、ワクチン接種)、併用免疫治療/細胞毒性方法(例えば、細胞毒性成分と結合した抗体)などの投与/適用と組合せることができる。
【0054】
別の実施態様においては、VPAまたはその誘導体は、VPA単独の局所適用によって皮膚腫瘍、基底細胞癌、扁平上皮細胞癌、角化棘細胞腫、ボーエン病、皮膚T細胞リンパ腫、光線性角化症、乾癬、魚鱗癬、座瘡、他の炎症性皮膚疾患を含む腫瘍性および非腫瘍性皮膚疾患における経口適用レチノイド活性を増強する。
【0055】
本発明は、皮膚/粘膜の癌、前悪性皮膚病変、および炎症の局所治療用に単独またはレチノイド、核内受容体リガンド、または5−FUなどの化学療法剤と組合せて使用されるバルプロ酸またはその誘導体を含有する医薬品の局所適用製剤を提供する。
【0056】
それゆえ、本発明の1つの態様は、皮膚/粘膜のさまざまなヒト癌、前悪性皮膚病変、または炎症の局所治療用、および日焼けの治療用に単独もしくはレチノイド、または5−フルオロウラシル(5−FU)と組合せて使用されるVPAまたはその誘導体を含有する局所適用製剤の使用である。したがって、各成分の単独の使用と比べた組合せ局所治療の抗腫瘍活性は増大しうるとともに―必要に応じて―かかる組合せ治療の個別成分の用量は、個別の薬剤単独による治療と関係した望ましくない副作用を減少させるために低下されうる。本発明はまた、上記の疾患の局所治療用の医薬品を製造するためのVPAまたはその誘導体の使用に関する。
【0057】
VPAの誘導体は、式I
【化2】

[式中、R1およびR2は独立して、場合により1個もしくは何個かのヘテロ原子を含んで成り、かつ置換されうる直鎖または分岐、飽和または不飽和、脂肪族C3-25炭化水素鎖であり、R3はヒドロキシル、ハロゲン、アルコキシ、または場合によりアルキル化アミノ基である]によって記載される炭素分岐カルボン酸またはカルボン酸誘導体として定義される。
【0058】
異なるR1およびR2残基はキラル化合物を生じさせる。通常、立体異性体の1つが他より強い催奇形効果を有し(ナウ(Nau)ら、1991年、Pharmacol.Toxicol.69、p.310−321)、催奇形異性体であるほど、より有効にPPARδを活性化する(ランペン(Lampen)ら、1999年)。したがって、この異性体はHDACをより強く阻害することが予想されうる(PCT/EP01/07704号)。本発明は、それぞれの化合物のラセミ混合物、具体的にはより活性の異性体を包含する。
【0059】
炭化水素鎖R1およびR2は、炭化水素鎖で炭素原子を置換する1個もしくは何個かのヘテロ原子(例えば、O、N、S)を含んで成りうる。これは、炭素基のものときわめて類似した構造物が、ヘテロ原子が対応する炭素基と同じ型のハイブリッド形成を有する場合にヘテロ原子基によって採用されうる事実による。
【0060】
R1およびR2は置換されうる。可能な置換基としては、ヒドロキシル基、アミノ基、カルボキシル基、およびアルコキシ基のほか、アリール基および複素環基が挙げられる。
【0061】
好ましくは、R1およびR2は独立して3〜10個、より好ましくは、4〜10個または5〜10個の炭素原子を含んでなる。R1およびR2は独立して飽和されており、または1つの二重結合または1つの三重結合を含んでなる。具体的には、側鎖(R1)の1つは、好ましくは、2および3位でsp1混成炭素原子、または同様の構造を生成するヘテロ原子を含有する。この側鎖は3個の炭素またはヘテロ原子を含んでなるが、長鎖もHDAC阻害分子を生成しうる。また、R2における芳香環またはヘテロ原子の包含は、HDACタンパク質の触媒部位が明らかに多種多様の結合分子を供給するためHDAC阻害活性を有する化合物を生成すると考えられる。催奇性VPA誘導体がHDAC阻害剤であるという所見により、以前に適切な抗てんかん薬とみなされていない化合物もHDAC阻害剤とみなされている(PCT/EP01/07704号)。具体的には、しかし排他的ではなく、R1としてプロピニル残基およびR2として7個もしくはそれ以上の炭素の残基を有する化合物が考えられている(ランペン(Lampen)ら、1999年)。
【0062】
好ましくは、基「COR3」はカルボキシル基である。カルボキシル基の誘導体化も潜在的なHDAC阻害活性を有する化合物の生成が考慮されなければならい。かかる誘導体は、ハロゲン化物(例えば、塩化物)、エステル、またはアミドでありうる。R3がアルコキシである場合、アルコキシ基は1〜25個、好ましくは、1〜10個の炭素原子を含んでなる。R3がモノ−またはジ−アルキル化アミノ基である場合、アルキル置換基は1〜25個、好ましくは、1〜10個の炭素原子を含んでなる。
【0063】
本発明によれば、式Iの化合物の医薬的に許容される塩も製剤用に使用されうる。本発明によれば、人体において式Iで定義される化合物に代謝され、または例えばエステル加水分解によって式Iで定義される化合物の放出をもたらす物質も使用されうる。
【0064】
特定の実施態様では、本発明は、ヒストンデアセチラーゼ活性を有する酵素の阻害剤として式Iに記載されたα−炭素分岐カルボン酸、またはその医薬的に許容される
塩の使用、および癌の局所治療におけるその使用に関し、式中R1は直鎖または分岐、飽和または不飽和、脂肪族C5-25炭化水素鎖であり、R2は独立して直鎖または分岐、飽和または不飽和、脂肪族C2-25炭化水素鎖、ただし、−CH2-CH=CH2、−CH2−C≡CHまたは−CH2−CH2−CH3ではなく、R1およびR2は場合により、ヒドロキシル基、アミノ基、カルボキシル基、アルコキシ基、アリール基、および/または複素環素で置換され、かつR3はヒドロキシルである。
【0065】
さらに別の実施態様では、本発明は、癌の局所治療用の式Iに記載されたα−炭素分岐カルボン酸、またはその医薬的に許容される塩に関し、式中R1は直鎖または分岐、飽和または不飽和、脂肪族C3-25炭化水素鎖であり、R2は独立して直鎖または分岐、飽和または不飽和、脂肪族C3-25炭化水素鎖であり、R1またはR2は炭化水素鎖で炭素原子を置換する1個もしくは何個かのヘテロ原子(例えば、O、N、S)を含んで成り、R1およびR2は場合により、ヒドロキシル基、アミノ基、カルボキシル基、アルコキシ基、アリール基、および/または複素環素で置換され、かつR3はヒドロキシルである。
【0066】
本発明のさらに別の実施態様では、R1およびR2はエステル基(−CO−O−)を含まない。R1およびR2は、ヘテロ原子O、N、またはSを含まない炭化水素鎖でありうる。
【0067】
本発明により使用される最も好ましい化合物は、VPAおよび/または4−ynVPAである。
【0068】
一般に、本発明は、さまざまなヒト疾患を治療する新しい可能性を提供する。出願人は、式Iの化合物のHDAC阻害および細胞分化誘導活性が単独で、または皮膚腫瘍および皮膚T細胞リンパ腫由来の細胞を含む腫瘍細胞の局所治療用のほか、前悪性皮膚病変用、および皮膚/粘膜の炎症性疾患用に既知でかつ臨床的に使用される治療薬と組合せて有効に使用されうることを見出した。組合せ治療は、それらだけで使用される対応する治療薬よりも患者において優れた治療効果を生むと考えられている。本発明の1つの目的は、皮膚に出現する癌の局所治療用に提示された化合物を使用する組合せ治療を提供することである。かかる組合せ治療により、例えば、必要される化学療法薬の治療量の減少がもたらされ、したがって、現在確認される、頻繁に使用される治療薬の部分的にきわめて重篤な副作用を制限しうる。
【0069】
本発明の態様としては、これに限定されないが、現在臨床的使用または臨床的開発における治療原理、例えば、
− 化学療法薬または細胞毒性薬(例えば、5−FU)
− 分化誘発薬剤(例えば、ビタミンD、ビタミンD誘導体、レチノイド、イミキモド(imiquimode)などの受容体結合剤)
− 放射線療法(例えば、エックス線またはガンマ線)
− 免疫学的方法(抗体療法、ワクチン接種)
− 併用免疫治療/細胞毒性法(例えば、細胞毒性成分を結合した抗体)
− 抗血管形成法
と、式Iの化合物との組み合わせを含む。
【0070】
製剤
本発明の組成物は、本発明の組成物が組込まれ、活性剤、および他の任意の活性物質が適切な濃度で皮膚に送達されることを可能にする皮膚科学的に許容される担体を約1%〜約99.9%を含んでなる。
【0071】
担体は、1つもしくはそれ以上の皮膚科学的に許容される固体、半固体、または液体、希釈剤、溶媒、増量剤などを含有しうる。担体は固体、半固体、または液体であってよい。好ましい担体は実質的に半固体である。担体はそれ自体、不活性であり、またはそれ自身の皮膚科学的利点を有しうる。担体の濃度は、選択された担体、活性剤および任意の成分の使用濃度によって変化しうる。
【0072】
適切な担体としては、皮膚科学的に許容される従来の、または他の既知の担体が挙げられる。担体はまた、本明細書に記載された必須成分と物理的および化学的に互換性があり、本発明の組成物と関係がある安定性、有効性、または他の使用利点を過度に損なうものであってはならない。本発明の組成物の好ましい成分は、通常の使用条件下に組成物の有効性を実質的に削減する相互作用がないような方法で混合されることが可能でなければならない。
【0073】
本発明で使用される担体の型は、組成物に所望の製品形態の型によって決まる。本発明において有用な局所組成物は、技術上周知であるような多種多様の製品形態に製造されうる。これらには、ローション、クリーム、ゲル、スティック、スプレー、軟膏、油、泡、粉末、およびペーストが含まれるが、これらに限定されない。これらの製品形態は、溶液、エアロゾル、乳剤、ゲル、固体、およびリポソームを含むが、これらに限定されないいくつかの型の担体を含んで成る。
【0074】
好ましい担体は、皮膚科学的に許容される、親水性希釈剤を含有する。本明細書で使用される「希釈剤」は、特定の材料が分散、溶解、または組込まれうる材料を含む。親水性希釈剤の限定されない例は、水、低級一価アルコール(例えば、C1−C4)、およびプロピレングリコール、ポリエチレングリコール(例えば、分子量200−600g/モル)、ポリプロピレングリコール(例えば、分子量425−2025g/モル)、グリセロール、ブチレングリコール、1,2,4−ブタントリオール、ソルビトールエステル、1,2,6−ヘキサントリオール、エタノール、イソプロパノール、ソルビトールエステル、ブタンジオール、エーテルプロパノール、エトキシル化エーテル、プロポキシル化エーテル、およびその組合せを含む低分子量グリコールおよびポリオールなど有機親水性希釈剤である。水が好ましい希釈である。組成物は、好ましくは、親水性希釈剤を約60%〜約99.99%含んでなる。
【0075】
本発明の溶液は、通常、皮膚科学的に許容される親水性希釈剤を含む。本発明において有用な溶液は、好ましくは、約60%〜約99.99%の親水性希釈剤を含む。
【0076】
本発明によるエアロゾルは、推進剤を上記の溶液に添加することによって形成されうる。例示的な推進剤にはクロロ−フッ化低分子量炭化水素が含まれる。エアロゾルは通常、スプレー式の製品として皮膚に適用される。
【0077】
好ましい担体は、親水性成分、例えば、水または他の親水性希釈剤を含んでなる親水性相、および疎水性成分、例えば、脂質、油、または油性物質を含んでなる疎水性相を含んでなる乳剤を含む。当業者には周知であるように、親水性相は疎水性相中に分散され、または逆もまた同様で、組成物の成分に応じてそれぞれ親水性または疎水性分散および連続相を形成する。乳剤技術においては、「分散相」という語は当業者には周知であり、この相が連続相に懸濁され、または連続相によって囲まれている小さな粒子または液滴として存在することを意味する。分散相は内相または不連続相としても知られている。乳剤は水中油型乳剤、またはシリコーン中水型乳剤など油中水型乳剤であり、または(例えば、三重または他の多相乳剤中に)これを含んで成りうる。水中油型乳剤は通常、約1%〜約50%の分散疎水性相を、かつ約1%〜約98%の連続親水性相を含んで成り、油中水型乳剤は通常、約1%〜約98%の分散親水性相を、約1%〜約50%の連続疎水性相を含んでなる。乳剤は、ゲルネットワークをも含んで成りうる。好ましい乳剤はさらに以下に記載されている。
【0078】
ローションおよびクリームを含むがこれらに限定されない本発明の局所組成物は、皮膚科学的に許容される皮膚軟化剤を含んで成りうる。かかる組成物は、好ましくは、皮膚軟化剤の約2%〜約50%を含有する。皮膚軟化剤は、皮膚をなめらかにする傾向があり、皮膚の平滑性および柔軟性を増大させ、皮膚の乾燥を防止または軽減し、かつ/または皮膚を保護する。皮膚軟化剤は通常、不水溶性、油性、またはロウ状材料である。皮膚軟化剤の非限定的な例が本明細書に記載されている。
【0079】
本発明によるローションおよびクリームは、好ましくは、溶液担体系および1つもしくはそれ以上の皮膚軟化剤を含んでなる。ローションは通常、約1%〜約20%、好ましくは、約5%〜約10%の皮膚軟化剤、約50%〜約90%、好ましくは、約60%〜約80%の水を含んでなる。クリームは通常、約5%〜約50%、好ましくは、約10%〜約20%の皮膚軟化剤、約45%〜約85%、好ましくは、約50%〜約75%の水を含んでなる。
【0080】
本発明の軟膏は、動物性もしくは植物性油または半固体炭化水素(油性)の単純担体基剤、水を吸収して乳剤を形成する吸収軟膏基剤、または水可溶性担体、例えば、水可溶性溶液担体を含んで成りうる。軟膏は、さらに増粘剤を含んで成りうる。例えば、軟膏は約2%〜約10%の皮膚軟化剤、かつ約0.1%〜約2%の増粘剤を含んで成りうる。軟化剤の非限定的な例が本明細書に記載されている。
【0081】
本発明の好ましい局所組成物は乳剤を含んでなる。本発明の乳剤は、以下の1つもしくはそれ以上を含んで成りうる。
【0082】
a)疎水性成分
本発明による乳剤は、脂質、油、油性、または他の疎水性成分を含んでなる疎水性相を含有する。本発明の組成物は、好ましくは、約1重量%〜約50重量%、好ましくは、約1重量%〜約30重量%、かつより好ましくは、約1重量%〜約10重量%の疎水性成分の組成物を含んでなる。疎水性成分は、動物、植物、または石油由来でありうるとともに、天然または合成(すなわち、人工)でありうる。好ましい疎水性成分は実質的に水不溶性、より好ましくは、基本的に水不溶性である。
【0083】
適切な疎水性成分の非限定的な例としては、以下より成る郡から選択されるものが挙げられる。
【0084】
(1)ワセリン液としても知られる鉱油は、石油から得られる液体炭化水素の混合物である。メルクインデックス(The Merck Index)、第10版、登録7048、p.1033(1983年)を参照。
【0085】
(2)ワセリンゼリーとしても知られる鉱油は、非直鎖固体炭化水素および高沸点液体炭化水素のコロイド系であり、液体炭化水素の大部分がミセル内に保持されている。メルクインデックス(The Merck Index)、第10版、登録7047、p.1033(1983年)、シンドラー(Schindler)、Drug.Cosmet.Ind.、89、p.36−37、p.76、p.78−80、p.82(1961年)を参照。
【0086】
(3)約7〜約40個の炭素原子を有する直鎖および分岐鎖炭化水素。これらの炭化水素材料の非限定的な例としては、ドデカン、イソドデカン、スクアラン、コレステロール、硬化ポリイソブチレン、ドコサン、ヘキサデカン、イソヘキサデカンが挙げられる。C7−C40分岐炭化水素、例えば、C13−C14イソパラフィンであるC7−C40イソパラフィンも有用である。
【0087】
(4)直鎖および分岐鎖材料ならびに芳香族誘導体を含むC1−C30カルボン酸およびC2−C30ジカルボン酸のC1−C30アルコールエステル(本明細書では疎水性成分、モノ−およびポリ−カルボン酸に関連して、直鎖、分岐鎖、およびアリールカルボン酸を含む)。非限定的な例としては、セバシン酸ジイソプロピル、アジピン酸ジイソプロピル、ミリスチン酸イソプロピル、パルミチン酸イソプロピル、パルミチン酸メチル、プロピオン酸ミリスチル、パルミチン酸2−エチルヘキシル、ネオペンタン酸イソデシル、マレイン酸ジ−2−エチルヘキシル、パルミチン酸セチル、ミリスチン酸ミリスチル、ステアリン酸ステアリル、ステアリン酸イソプロピル、ステアリン酸メチル、ステアリン酸セチル、ベヘン酸ベヘニル(behenyl behenrate)、マレイン酸ジオクチル、セバシン酸ジオクチル、アジピン酸ジイソプロピル、オクタン酸セチル、ジリノール酸ジイソプロピルが挙げられる。
【0088】
(5)C1−C30カルボン酸のモノ−、ジ−、およびトリ−グリセリド、例えば、カプリル/トリカプリン酸グリセリル、PEG−6カプリル/トリカプリン酸グリセリル、 PEG−8カプリル/トリカプリン酸グリセリル。
【0089】
(6)C1−C30カルボン酸のアルキレングリコールエステル、例えば、エチレングリコールモノ−およびジ−エステル、およびC1−C30カルボン酸のプロピレングリコールモノ−およびジ−エステル、例えば、ジステアリン酸エチレングルコール。
【0090】
(7)前記材料のプロポキシル化およびエトキシル化誘導体。
【0091】
(8)糖および関連材料のC1−C30モノ−およびポリ−エステル。これらのエステルは糖またはポリオール部分および1つもしくはそれ以上のカルボン酸部分由来である。構成酸および糖に応じて、これらのエステルは室温下に液体または固体形態のいずれかである。液体エステルの例としては、テトラオレイン酸グルコース、大豆油脂肪酸のグルコーステトラエステル(不飽和)、混合大豆油脂肪酸のマンノーステトラエステル、オレイン酸のガラクトーステトラエステル、リノール酸のアラビノーステトラエステル、テトラリノール酸キシロース、ペンタオレイン酸ガラクトース、テトラオレイン酸ソルビトール、不飽和大豆油脂肪酸のソルビトールヘキサエステル、ペンタオレイン酸キシリトール、テトラオレイン酸ショ糖、ペンタオレイン酸ショ糖、ヘキサオレイン酸ショ糖、ヘパトオレイン酸ショ糖、オクタオレイン酸ショ糖、およびその混合物が挙げられる。固体エステルの例としては、カルボン酸エステル部分が1:2のモル比でパルミトレイン酸とアラキジン酸であるソルビトールヘキサエステル、カルボン酸エステル部分が1:3のモル比でリノール酸とベヘン酸であるラフィノースのオクタエステル、エステル化カルボン酸部分が、3:4のモル比でヒマワリ種子油脂肪酸とリグノセリン酸であるマルトースのヘプタエステル、エステル化カルボン酸部分が、2:6のモル比でオレイン酸とベヘン酸であるショ糖のオクタエステル、およびエステル化カルボン酸部分が、1:3:4のモル比でラウリン酸とリノール酸とベヘン酸であるショ糖のオクタエステルが挙げられる。好ましい固体材料が、エステル化の程度が7−8であり、不飽和:ベヘル酸のモル比が1:7〜3:5である脂肪酸部分がC18モノ−および/またはジ−不飽和およびベヘン酸であるショ糖ポリエステルである。特に好ましい固体糖ポリステルが、約7個のベヘン脂肪酸部分と約1個のオレイン酸部分が分子中にあるショ糖のオクタエステルである。他の材料としては、ショ糖の綿実油または大豆油脂肪酸エステルが挙げられる。
【0092】
(9)有機ポリシロキサン油。有機ポリシロキサン油は、揮発性、不揮発性、または揮発性と不揮発性のシリコーンの混合物でありうる。この文脈において使用される「不揮発性」という語は、周囲条件下に液体であるシリコーンを指す。この文脈において使用される「揮発性」という語は、他のすべてのシリコーン油を指す。適切な有機ポリシロキサンが、広範囲の揮発度および粘度にわたる多種多様のシリコーンから選択されうる。不揮発性ポリシロキサンが好ましい。適切な有機ポリシロキサン油の例としては、ポリアルキルシロキサン、環状ポリアルキルシロキサン、およびポリアルキルアリールシロキサンが挙げられる。
【0093】
ここでの使用に好ましいのは、ポリアルキルシロキサン、アルキル置換ジメチコーン、シクロメチコーン、トリメチルシルオキシシリケート、ジメチコノール、ポリアルキルアリールシロキサン、およびその混合物から成る群より選択される有機ポリシロキサンである。ここでの使用により好ましいのは、ポリアルキルシロキサンおよびシクロメチコーンである。ポリアルキルシロキサンの中でも好ましいのは、米国特許第5,968,528号明細書に記載されているジメチコーンでである。
【0094】
(10)植物性油および硬化植物性油。植物性油および硬化植物性油の例としては、ヒワマリ油、ヒマシ油、ココナッツ油、綿実油、ニシン油、パーム核油、ヤシ油、ピーナッツ油、大豆油、菜種油、アマニ油、ぬか油、松根油、ゴマ油、ヒマワリ種子油、硬化ベニバナ油、硬化ヒマシ油、硬化ココナッツ油、硬化綿実油、硬化ニシン油、硬化パーム核油、硬化ヤシ油、硬化ピーナッツ油、硬化大豆油、硬化菜種油、硬化アマニ油、硬化ぬか油、硬化ゴマ油、硬化ヒマワリ種子油、およびその混合物が挙げられる。
【0095】
(11)動物油脂、例えば、ラノリンまたはその誘導体、タラ肝油。
【0096】
(12)ポリプロピレングリコールのC4−C20アルキルエーテル、ポリプロピレングリコールのC1−C20カルボン酸エステル、およびジ−C8−C30アルキルエーテルもまた有用である。これらの材料の非限定的な例としては、PPG−14ブチルエーテル、PPG−15ステアリルエーテル、ジオクチルエーテル、ドデシルオクチルエーテルおよびその混合物が挙げられる。
【0097】
b)親水性成分
本発明の乳剤は、親水性成分、例えば、水または他の親水性希釈剤をも含んでなる。したがって、親水性相は、水、または水と1つもしくはそれ以上の水可溶性または分散可能な成分の組合せを含んで成りうる。水を含んでなる親水性成分が好ましい。
【0098】
(c)他の成分
本発明の乳剤および他の局所組成物は、本明細書に記載されているものなどさまざまな他の成分を含んで成る。当業者によって理解されるように、組成物中の成分の親水性に応じて、所定の成分が主に親水性相または疎水性相のいずれかに分散する。
【0099】
本発明の乳剤としては、好ましくは、乳化剤、界面活性剤、構造化剤、および増粘剤から選択される1つもしくはそれ以上の化合物が挙げられる。
【0100】
(1)乳化剤/界面活性剤
乳剤は、連続相内の不連続相を分散および懸濁するのに一般に役立つ乳化剤および/または界面活性剤を含有しうる。多種多様のかかる薬剤が使用されうる。選択された薬剤が化学的および物理的に組成物の基本的な成分と互換性があり、かつ所望の分散特性を示すという条件で、既知または従来の乳化剤/界面活性剤を組成物において使用することができる。適切な薬剤としては、非シリコーン含有乳化剤/界面活性剤、シリコーン乳化剤/界面活性剤、およびその混合物が挙げられる。
【0101】
好ましい実施態様においては、組成物は、親水性乳化剤または界面活性剤を含んでなる。本発明の組成物は、好ましくは、約0.05%〜約5%、より好ましくは、約0.05%〜約1%の少なくとも1つの親水性界面活性剤を含んでなる。
【0102】
好ましい親水性界面活性剤は非イオン界面活性剤から選択される。ここで有用である非イオン界面活性剤は中でも長鎖アルコールの縮合生成物として広範囲に定義されているもの、例えば、糖またはでんぷんポリマーを有する、すなわちグリコシドであるC8−30アルコールである。アルキル基がそれに由来しうる長鎖アルコールの例としては、デシルアルコール、セチルアルコール、ステアリルアルコール、ラウリルアルコール、ミリスチルアルコール、オレイルアルコールなどが挙げられる。これらの界面活性剤の市販の例としては、デシルポリグルコシド(APG 325 CSとしてヘンケル(Henkel)から入手可能)およびラウリルポリグルコシド(APG 600 CSおよび625 CSとしてヘンケル(Henkel)から入手可能)が挙げられる。
【0103】
他の有用な非イオン界面活性剤としては、脂肪酸とのアルキレンオキシドの縮合生成物(すなわち、脂肪酸のアルキレンオキシドエステル)が挙げられる。他の非イオン界面活性剤は、脂肪アルコール酸とのアルキレンオキシドの縮合生成物(すなわち、脂肪アルコールのアルキレンオキシドエーテル)である。さらに他の非イオン界面活性剤は、脂肪酸および脂肪アルコールとのアルキレンオキシドの縮合生成物である[すなわち、ここでポリアルキレンオキシド部分は、一端で脂肪酸によりエステル化されており、もう一端で脂肪アルコールによりエーテル化(すなわち、エーテル連鎖によって結合)されている]。これらアルキレンオキシド由来非イオン界面活性剤の例としては、セテス−6、セテス−10、セテス−12、セテアレス−6、セテアレス−10、セテアレス−12、ステアレス−6、ステアレス−10、ステアレス−12、PEG−6ステアリン酸、PEG−10ステアリン酸、PEG−100ステアリン酸、PEG−12ステアリン酸、PEG−20ステアリン酸グリセリル、PEG−80グリセリル牛脂脂肪酸、PEG−10ステアリン酸グリセリル、PEG−30グリセリルココエート、PEG−80グリセリルココエート、PEG−200グリセリル牛脂脂肪酸、PEG−8ジラウリン酸、PEG−10ジステアリン酸塩、およびその混合物が挙げられる。
【0104】
さらに他の有用な非イオン界面活性剤としては、ポリヒドロキシ脂肪酸アミド界面活性剤が挙げられる。
【0105】
非イオン界面活性剤の中で好ましいのは、ステアレス−21、セテアレス−20、セテアレス−12、ショ糖ココエート、ステアレス−100、PEG−100ステアリン酸塩、およびその混合物から成る群より選択されるものである。
【0106】
この点で使用に適切な他の非イオン界面活性剤としては、糖エステルおよびポリエステル、アルコキシル化糖エステルおよびポリエステル、C1−C30脂肪アルコールのC1−C30脂肪酸エステル、C1−C30脂肪アルコールのC1−C30脂肪酸エステルのアルコキシル化誘導体、C1−C30脂肪アルコールのアルコキシル化エーテル、C1−C30脂肪酸のポリグリセリルエステル、ポリオールのC1−C30エステル、ポリオールのC1−30エーテル、アルキルリン酸塩、ポリオキシアルキレン脂肪エーテルリン酸塩、脂肪酸アミド、乳酸アシル、およびその混合物が挙げられる。これら非シリコン含有乳化剤の非限定的な例としては、ポリエチレングリコール20ソルビタンモノラウレート(ポリソルベート20)、ポリエチレングリコール5大豆ステロール、ステアレス−20、セテアレス−20、PPG−2メチルグルコースエーテルジステアリン酸塩、セテス−10、ポリソルベート80、リン酸セチル、リン酸セチルカリウム、ジエタノールアミンリン酸セチル、ポリソルベート60、ステアリン酸グリセリル、ポリオキシエチレン20トリオレイン酸ソルビタン(ポリソルベート85)、ソルビタンモノラウレート、ポリオキシエチレン4ラウリルエーテルステアリン酸ナトリウム、イソステアリン酸ポリグリセリル−4、ラウリン酸ヘキシル、PPG−2ジステアリン酸メチルグルコースエーテル、PEG−100ステアリン酸塩、およびその混合物が挙げられる。
【0107】
ここで有用な他の乳化剤は、ソルビタンまたはソルビトール脂肪酸エステルおよびショ糖脂肪酸エステルの混合物に基づく脂肪酸エステル混合物である。好ましい脂肪酸エステル乳化剤は、ソルビタンまたはソルビトールC16−C20脂肪酸エステルのショ糖C10−C16脂肪酸エステル、特にソルビタンステアリン酸およびショ糖ココエートとの混合物である。これは、アルラトン(Arlatone)2121の商品名でICIから市販されている。
【0108】
ここで有用な親水性界面活性剤は、代わりとして、または追加として、技術上既知である多種多様の陽イオン、陰イオン、両性イオン、および両性界面活性剤のいずれかを含む。例えば、MaCutcheon’s Detergents and Emulsifiers、North Americann版(1986年)、アルレード出版社(Allured Publishing Corporation)刊を参照。
【0109】
ここで有用な例示的な陽イオン界面活性剤としては、米国特許第5,968,528号明細書に開示されているものが挙げられる。ここで有用な陽イオン界面活性剤としては、四級アンモニウム塩、およびアミノ−アミドなど陽イオンアンモニウム塩が挙げられる。
【0110】
多種多様の陰イオン界面活性剤もここでは有用である。例えば、米国特許第5,968,528号明細書を参照。陰イオン界面活性剤の非限定的な例としては、イセチオン酸アルコイル(例えば、C12−C30)、硫酸アルキルおよびアルキルエーテル、およびその塩、リン酸アルキルおよびアルキルエーテル、およびその塩、タウリン酸アルキルメチル(例えば、C12−C30)、および脂肪酸のせっけん(例えば、アルカリ金属塩、例えば、ナトリウムまたはカリウム塩)が挙げられる。
【0111】
両性イオンおよび両性界面活性剤もここで有用である。本発明の組成物において使用されうる両性イオンおよび両性界面活性剤の例は、脂肪族ラジカルが直鎖または分岐鎖でありうるとともに、脂肪族置換基の1つが約8〜約22個の炭素原子(好ましくは、C8−C18)を含有し、1つが陰イオン水可溶化基、例えば、カルボキシ、スルホン酸塩、硫酸塩、リン酸塩、またはホスホン酸塩を含有する、脂肪族第二級および第三級アミンの誘導体として広範囲に記載されているものである。例えば、アルキルイミノ酢酸塩、およびイミノジアルカン酸塩およびアミノアルカン酸塩、イミダゾリニウム、およびアンモニウム誘導体である。他の適切な両性イオンおよび両性界面活性剤は、ベタイン、スルタイン、ヒドロキシスルタイン、アルキルサルコシン酸塩(例えば、C12−C30)、およびアルカノイルサルコシン酸塩よりなる群から選択されるものである。
【0112】
本発明の好ましい乳剤としては、乳化剤または界面活性剤を含有するシリコーンが挙げられる。多種多様のシリコーン乳化剤がここで有用である。これらのシリコーン乳化剤は通常、当業者にはシリコーン界面活性剤としても知られる有機修飾された有機ポリシロキサンである。有用なシリコーン乳化剤としては、ジメチコーンコポリオールが挙げられる。これらの材料は、ポリエチレンオキシド鎖、ポリプロピレンオキシド鎖、これらの鎖の混合物、およびエチレンオキシドおよびプロピレンオキシド由来の部分を含有するポリエーテル鎖などポリエーテル側鎖を含むように修飾されているポリジメチルシロキサンである。他の例としては、アルキル修飾ジメチコーンコポリオール、すなわち、C2−C30ペンダント側鎖を含有する化合物が挙げられる。さらに他の有用なジメチコーンコポリオールとしては、さまざまな陽イオン、陰イオン、両性イオン、および両性ペンダント部分を有する材料が挙げられる。
【0113】
ここで乳化剤として有用なジメチコーンコポリオールおよび他のシリコーン界面活性剤の非限定的な例としては、ペンダントポリエチレンオキシド側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダントポリプロピレンオキシド側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダント混合ポリエチレンオキシドおよびポリプロピレンオキシド側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダント混合ポリ(エチレン)(プロピレン)オキシド側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダントオルガノベタイン側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダントカルボン酸側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダント四級アンモニウム側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、かつペンダントC2−C30直鎖、分岐、または環状アルキル部分を含有する前記コポリマーの別の修飾も挙げられる。ダウコーニング社(Dow Corning Corporation)によって販売されているここで有用な市販のジメチコーンコポリオールの例は、ダウコーニング(Dow Corning)(登録商標)190、193、Q2−5220、2501ロウ(Wax)、2−5324流体、および3225Cである(後者の材料は、シクロメチコーンとの混合物として販売されている)。セチルジメチコーンコポリオールは、ポリグリセリル−4イソステアリン酸(および)ラウリン酸ヘキシルとの混合物として市販されており、ABIL(登録商標)WE−09の商品名で販売されている(ゴールドシュミット(Goldschmidt)から入手可能)。セチルジメチコーンコポリオールは、ラウリン酸ヘキシル (および)ポリグリセリル−3オレイン酸(および)セチルジメチコーンとの混合物としても市販されており、ABIL(登録商標)WS−08の商品名で販売されている(やはりゴールドシュミット(Goldschmidt)から入手可能)。ジメチコーンコポリオールの他の非限定的な例としては、ラウリルジメチコーンコポリオール、ジメチコーンコポリオール酢酸塩、ジメチコーンコポリオールアジピン酸塩、ジメチコーンコポリオールアミン、ジメチコーンコポリオールベヘン酸塩、ジメチコーンコポリオールブチルエーテル、ジメチコーンコポリオールヒドロキシステアリン酸塩、ジメチコーンコポリオールイソステアリン酸塩、ジメチコーンコポリオールラウリン酸塩、ジメチコーンコポリオールメチルエーテル、ジメチコーンコポリオールリン酸塩、およびジメチコーンコポリオールステアリン酸塩も挙げられる。
【0114】
ここで有用なジメチコーンコポリオール乳化剤が、例えば、米国特許第5,968,528号明細書に記載されている。
【0115】
(2)構造化剤
この組成物、および特にこの乳剤は、構造化剤を含有しうる。構造化剤は特に本発明の水中油型乳剤において好ましい。理論によって限定されることなく、構造化剤は、組成物の安定性に寄与するレオロジー特性を組成物に提供するのに役立つ。例えば、構造化剤は、液晶ゲルネットワークの形成を補助する傾向がある。構造化剤は、乳化剤または界面活性剤としても機能しうる。本発明の好ましい組成物は、約1%〜約20%、より好ましくは、約1%〜約10%、最も好ましくは、約2%〜約9%の1つもしくはそれ以上の構造化剤を含んでなる。
【0116】
好ましい構造化剤は、約1〜約8個のHLBを有し、かつ少なくとも約45℃の融点を有するものである。適切な構造化剤は、飽和C14〜C30脂肪アルコール、約1〜約5モルのエチレンオキシドを含有する飽和C16〜C30脂肪アルコール、飽和C16〜C30ジオール、飽和C16〜30モノグリセロールエーテル、飽和C16〜C30ヒドロキシ脂肪酸、C14〜C30ヒドロキシル化および非ヒドロキシル化飽和脂肪酸、C14〜C30飽和エトキシル化脂肪酸、アミン、および約1〜約5モルのエチレンオキシドジオールを含有するアルコール、少なくとも40%のモノグリセリド含量を有するC14〜C30飽和グリセリルモノエステル、約1〜約3個のアルキル基および約2〜約3個の飽和グリセロール単位を有するC14〜C30飽和ポリグリセロールエステル、C14〜C30グリセリルモノエーテル、C14〜C30ソルビタンモノ/ジエステル、約1〜約5モルのエチレンオキシドを有するC14〜C30飽和エトキシル化ソルビタンモノ/ジエステル、C14〜C30飽和メチルグルコシドエステル、C14〜C30飽和ショ糖モノ/ジエステル、約1〜約5モルのエチレンオキシドを有するC14〜C30飽和エトキシル化メチルグルコシドエステル、少なくとも約45℃の融点を有する平均1〜2個のグルコース単位を有するC14〜C30飽和ポリグルコシド、およびその混合物から成る群より選択されるものである。
【0117】
本発明の好まし構造化剤は、ステアリン酸、パルミチン酸、ステアリルアルコール、セチルアルコール、ベヘニルアルコール、ステアリン酸、パルミチン酸、平均約1〜約5個のエチレンオキシド単位を有するステアリルアルコールのポリエチレングリコールエーテル、平均約1〜約5個のエンチレンオキシド単位を有するセチルアルコールのポリエチレングリコールエーテル、およびその混合物から成る群より選択される。本発明のより好ましい構造化剤は、ステアリルアルコール、セチルアルコール、ベヘニルアルコール、平均約2個のエチレンオキシド単位(ステアレス−2)を有するステアリルアルコールのポリエチレングリコールエーテル、平均約2個のエチレオキシド単位を有するセチルアルコールのポリエチレングリコールエーテル、およびその混合物から成る群より選択される。さらにより好ましい構造化剤は、ステアリン酸、パルミチン酸、ステアリルアルコール、セチルアルコール、ベヘニルアルコール、ステアレス−2、およびその混合物から成る群より選択される。
【0118】
(3)増粘剤(増粘剤およびゲル化剤を含む)
本発明の組成物はまた、好ましくは、約0.1%〜約5%、より好ましくは、約0.1%〜約3%、および最も好ましくは、約0.25%〜約2%の増粘剤を含んで成る。
【0119】
増粘剤の非限定的なクラスとしては、以下のから成る群より選択される。
【0120】
(i)カルボン酸ポリマー
これらのポリマーは、アクリル酸、置換アクリル酸、およびこれらアクリル酸および置換アクリル酸の塩およびエステル由来の1つもしくはそれ以上のモノマーを含有する架橋化合物であり、ここで架橋剤は2つもしくはそれ以上の炭素−−炭素二重結合を含み、多価アルコール由来である。好ましいカルボン酸ポリマーは、2つの一般的な型である。第1の型のポリマーは、アクリル酸モノマーまたはその誘導体の架橋ホモポリマーである(例えば、ここでアクリル酸は、C1−4アルキル、−−CN、−−COOH、およびその混合物から成る群より独立して選択される2個および3個の炭素位置で置換基を有する)。第2の型のポリマーは、アクリル酸モノマーまたはその誘導体(前文に記載したように)、短鎖アルコール(すなわち、C1−4)アクリレートエステルモノマーまたはその誘導体(例えば、ここでエステルのアクリル酸部分は、C1−4アルキル、−−CN、−−COOH、およびその混合物から成る群より独立して選択される2個および3個の炭素位置で置換基を有する)、およびその混合物から成る群より選択される第1のモノマー、および長鎖アルコール(すなわち、C8−40)アクリレートエステルモノマーまたはその誘導体(例えば、ここでエステルのアクリル酸部分は、C1−4アルキル、−−CN、−−COOH、およびその混合物から成る群より独立して選択される2個および3個の炭素位置で置換基を有する)である第2のモノマーを有する架橋コポリマーである。これら2つの型のポリマーの組合せもここで有用である。
【0121】
第1の型の架橋ホモポリマーにおいて、モノマーは、好ましくは、アクリル酸、メタクリル酸、エタクリル酸、およびその混合物から成る群より選択され、アクリル酸が最も好ましい。第2の型の架橋コポリマーにおいては、アクリル酸モノマーまたはその誘導体は、好ましくは、アクリル酸、メタクリル酸、エタクリル酸、およびその混合物から成る群より選択され、アクリル酸、メタクリル酸、およびその混合物が最も好ましい。短鎖アルコールアクリレートエステルモノマーまたはその誘導体は、好ましくは、C1−4アルコールアクリレートエステル、C1−4アルコールメタクリレートエステル、C1−4アルコールエタクリレートエステル、およびその混合物から成る群より選択され、C1−4アルコールアクリレートエステル、C1−4アルコールメタクリレートエステル、およびその混合物が最も好ましい。長鎖アルコールアクリレートエステルモノマーは、C8−40アルキルアクリレートエステルから選択され、C10−30アルキルアクリレートエステルが好ましい。
【0122】
これらの型のポリマーの両方における架橋剤は、分子当り2個以上のアルケニルエーテル基を含有する多価アルコールのポリアルケニルポリエーテルであり、ここで親多価アルコールは少なくとも3個の炭素原子および少なくとも3個のヒドロキシル基を含有する。好ましい架橋剤は、ショ糖のアリルエーテルおよびペンタエリトリトールのアリルエーテル、およびその混合物から成る群より選択されるものである。
【0123】
ここで有用な第1の型の市販のホモポリマーの例としては、ショ糖またはペンタエリトリトールのアリルエーテルを有するアクリル酸架橋のホモポリマーであるカルボマーである。カルボマーは、カルボポール(Carbopol)(登録商標)900シリーズとしてB.F.グッドリッチ(Goodrich)から入手可能である(例えば、カルボポール(Carbopol)(登録商標)954)。ここで有用な第2の型の市販のコポリマーの例としては、アクリル酸、メタクリル酸、またはその短鎖(すなわち、C1−4アルコール)エステルの1つのモノマーの1つもしくはそれ以上を有するC10−30アルキルアクリレートのコポリマーであり、ここで架橋剤はショ糖またはペンタエリトリトールのアリルエーテルである。これらのコポリマーは、アクリレート/C10−30アルキルアクリレートクロスポリマーとして知られており、カルボポール(Carbopol)(登録商標)1342、カルボポール(Carbopol)(登録商標)1382ペムレン(Pemulen)TR−1、およびペムレン(Pemulen)TR−2として、B.F.グッドリッチ(Goodrich)から市販されている。言い換えれば、ここで有用なカルボン酸ポリマー増粘剤の例は、カルボマー、アクリレート/C10−30アルキルアクリレートクロスポリマー、およびその混合物から成る群より選択されるものである。
【0124】
(ii)架橋ポリアクリレートポリマー
増粘剤またはゲル化剤として有用な架橋ポリアクリレートポリマーとしては、陽イオンおよび非イオンポリマーが挙げられ、陽イオンポリマーが一般に好ましい。適切な架橋ポリアクリレートポリマーの非限定的な例は、米国特許第5,968,528号明細書に開示されている。
【0125】
(iii)ポリアクリルアミドポリマー
ポリアクリルアミドポリマー、特に、置換分岐または非分岐ポリマーを含む非イオンポリアクリルアミドポリマーもここで有用である。これらのポリマーは、1個または2個のアルキル基(好ましくは、C1〜C5)で置換されていない、または置換されているアクリルアミドおよびメタクリルアミドを含むさまざまなモノマーから形成されうる。アミド窒素が置換されていない、または1個または2個のC1〜C5アルキル基(好ましくは、メチル、エチル、またはプロピル)で置換されているアクリレートアミドおよびメタクリレートアミドモノマー、例えば、アクリルアミド、メタクリルアミド、N−メタクリルアミド、N−メチルメタクリルアミド、N,N−ジメチルメタクリルアミド、N−イソプロピルアクリルアミド、N−イソプロピルメタクリルアミド、およびN,N−ジメチルアクリルアミドが好ましい。これらのポリマーは、約1,000,000より大きい、好ましくは、約1,5000,000よりも大きい、かつ約30,000,000までの分子量を有する。これらのポリアクリルアミドポリマーの中で最も好ましいのは、CTFAで表示されるポリアクリルアミドである非イオンポリマーおよびイソパラフィンおよびラウレス−7(laureth−7)であり、これはセピゲル(Sepigel)305の商品名でセピック社(Seppic Corporation)(ニュージャージー州(N.J.)、フェアフィールド(Fairfield))から入手可能である。
【0126】
ここで有用な他のポリアクリルアミドポリマーとしては、アクリル酸および置換アクリル酸を有するアクリルアミドおよび置換アクリルアミドの多ブロックコポリマーが挙げられる。これらの多ブロックコポリマーの市販の例としては、リポケミカルズ社(Lipo Chemicals,Inc.)(ニュージャージー州(N.J.)、パターソン(Patterson))のハイパン(Hypan)SR150H、SS500V、SS500W、SSSA100Hが挙げられる。
【0127】
(iv)多糖類
多種多様の多糖類がここで有用である。「多糖類」によって、繰り返し糖(すなわち、糖質)単位の骨格を含有するゲル化剤が意味される。多糖類ゲル化剤の非限定的な例としては、セルロース、カルボキシメチルヒドロキシエチルセルロース、セルロースアセテートプロピオン酸カルボキシレート、ヒドロキシエチルセルロース、ヒドロキシエチルエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、メチルヒドロキシエチルセルロース、微結晶性セルロース、硫酸ナトリウムセルロース、およびその混合物から成る群より選択されるものである。ここではアルキル置換セルロースも有用である。これらのポリマーにおいては、セルロースポリマーのヒドロキシ基は、ヒドロキシアルキル化(好ましくは、ヒドロキシエチル化またはヒドロキシプロピル化)されてヒドロキシアルキル化セルロースを形成し、これは次いでエーテル連鎖によりC10−C30直鎖または分岐鎖アルキル基で修飾される。通常、これらのポリマーは、ヒドロキシアルキルセルロースを有するC10−C30直鎖または分岐鎖アルコールのエーテルである。ここで有用なアルキル基の例としては、ステアリル、イソステアリル、ラウリル、ミリスチル、セチル、イソセチル、ココイル(すなわち、ココナッツ油のアルコール由来のアルキル基)、パルミチル、オレイル、リノレイル、リノレニル、リシノレイル、ベヘニル、およびその混合物から成る群より選択されるものが挙げられる。中でも好ましいアルキルヒドロキシアルキルセルロースエーテルは、CTFAで表示される材料であるセチルヒドロキシエチルセルロースであり、これはセチルアルコールおよびヒドロキシエチルセルロースのエーテルである。この材料は、ナトロソール(Natrosol)(登録商標)CSプラスの商品名でアクアロン社(Aqualon Corporation)から市販されている。
【0128】
v)ゴム
ここで有用な他の追加の増粘化およびゲル化剤としては、主に天然源由来である材料が挙げられる。これらのゲル化剤ゴムの非限定的な例としては、アカシア、寒天、アルギン、アルギン酸、アルギン酸アンモニウム、アミロペクチン、アルギン酸カルシウム、カラギーナンカルシウム、カルニチン、カラギーナン、デキストリン、ゼラチン、ゲランゴム、グアーガム、グアーヒドロキシプロピルトリモニウムクロリド、ヘクトライト、ヒアルロン酸、ケイ酸、ヒドロキシプロピルキトサン、ヒドロキシプロピルグアー、カラヤゴム、昆布、ローカストビーンゴム、納豆ゴム、アルギン酸カリウム、カリウムカラギーナン、アルギン酸プロピレングリコール、スクレロチウムゴム、カルボイキシメチルデキストランナトリウム、カラギーナンナトリウム、トラガカントゴム、キサンタンガム、およびその混合物から成る群より選択される材料が挙げられる。
【0129】
(vi)架橋ビニルエーテル/無水マレイン酸コポリマー
ここで有用な他の追加の増粘化およびゲル化剤としては、アルキルビニルエーテルおよび無水マレイン酸の架橋コポリマーが挙げられる。
【0130】
(vii)架橋ポリ(N−ビニルピロリドン)
追加の増粘化およびゲル化剤としてここで有用な架橋ポリビニル(N−ピロリドン)。これらのゲル化剤は通常、約2〜約12個の炭素原子を含有する末端ジオールのジビニルエーテルおよびジアリルエーテル、約2〜約600単位を含有するポリエチレングリコールのジビニルエーテルおよびジアリルエーテル、約6〜約20個の炭素原子を有するジエン、ペンタエリトリトールのジビニルベンゼン、ビニル、およびアリルエーテルなどから成る群より選択される架橋剤約0.25〜約1重量%を含有する。
【0131】
本発明の好ましい組成物としては、カルボン酸ポリマー、架橋ポリアクリレートポリマー、ポリアクリルアミドポリマー、およびその混合物から成る群より選択され、より好ましくは、架橋ポリアクリレートポリマー、ポリアクリルアミドポリマー、およびその混合物から成る群より選択される増粘剤が挙げられる。
【0132】
任意の成分
本発明の局所組成物は多種多様の任意の成分を含んで成りうるが、かかる任意の成分は物理的および化学的に本明細書に記載された基本的な成分と互換性があり、本発明の組成物と関係した安定性、有効性、または他の使用利点を過度に損なうことがないことを条件とする。任意の成分を、本組成物の担体中に分散、溶解などすることができる。
【0133】
任意の成分としては、エステ剤および活性剤が挙げられる。例えば、組成物としては、本発明の基本的な成分に加えて、吸収剤(粘土など油吸収剤およびポリマー吸収剤を含む)、研磨剤、アンチケーキング剤、消泡剤、抗菌剤(例えば、微生物を破壊し、微生物の成長を阻止し、または微生物の病原性作用を阻止することが可能であり、例えば、座瘡の制御および/または局所組成物の保存に有用である化合物)、結合剤、生物学的添加剤、緩衝剤、充填剤、化学添加剤、化粧殺生剤、変性剤、化粧収斂剤、薬剤収斂剤、外用鎮痛薬、被膜剤、保湿剤、乳白剤、芳香剤、香料、顔料、着色剤、精油、皮膚知覚剤(skin sensate)、皮膚軟化剤、皮膚緩和剤、皮膚治療薬、pH調整剤、可塑剤、防腐剤、防腐増強剤、推進剤、還元剤、皮膚調整剤、皮膚透過増強剤、皮膚保護剤、溶媒、懸濁剤、乳化剤、増粘剤、可溶化剤、組成物の被膜特性および実質性を補助するためのポリマー(例えば、エイコセンス(eicosense)およびビニルピロリドンのコポリマーであり、その例はGAFケミカル社(Chemical Corp)からガネックス(Ganex)(登録商標)V−220として入手可能)、ロウ、日焼け止め剤(sunscreen)、日焼け止め剤(sunblock)、紫外線吸収剤または散乱剤、サンレスタンニング剤、抗酸化剤および/またはラジカルスカベンジャー、キレート化剤、金属イオン封鎖剤、抗座瘡剤、抗炎症剤、抗男性ホルモン物質、脱毛剤、落屑剤/エクスフォリアント、有機ヒドロキシ酸、ビタミン、およびその誘導体(ビタミンCおよびリン酸アスコルビルなど水分散性または可溶性ビタミンを含む)、コラーゲン産生を刺激する化合物、および天然抽出物を挙げることができる。他のかかる材料は技術上既知である。かかる材料の非排他的な例は、Pharmaceutical Dosage Forms−Disperse Systems、リーバーマン(Lieberman)、リーガー・アンド・バンカー(Riger & Banker)、第1巻(1988年)および第2巻(1989年)、または米国特許第5,968,528号明細書に記載されている。
【0134】
本発明の組成物は、皮膚軟化剤を含んで成りうる。皮膚軟化剤は、以下のクラスの1つもしくはそれ以上から選択されうる。すなわち、植物性および動物性油、およびヒマシ油、カカオバター、ベニバナ油、綿実油、トウモロコシ油、オリーブ油、タラ肝油、アーモンド油、アボガド油、ヤシ油、ゴマ油、スクアレン、キクイ(kikui)油、および大豆油を含むがこれらに限定されないトリグリセリド、アセチル化モノグリセリドなどアセトグリセリドエステル、エトキシル化グリセリルモノステアリン酸などエトキシル化グリセリド、メチル、イソプロピル、およびラウリン酸ヘキシル、ラウリン酸イソヘキシル、パルミチン酸イソヘキシル、パルミチン酸イソプロピル、パルミチン酸メチル、オレイン酸デシル、オレイン酸イソデシル、ステアリン酸ヘキサデシル、ステアリン酸デシル、イソステアリン酸イソプロピル、イソステアリン酸メチル、アジピン酸ジイソプロピル、アジピン酸ジイソヘキシル、アジピン酸ジヘキシルデシル、セバシン酸ジイソプロピル、乳酸ラウリル、乳酸ミリスチル、乳酸セチルなどの脂肪酸のブチルエステルを含むがこれらに限定されない、10〜20個の炭素原子を有する脂肪酸のアルキルエステル、ミリスチン酸オレイル、ステアリン酸オレイル、およびオレイン酸オレイルなど10〜20個の炭素原子を有する脂肪酸のアルケニルエステル、ペラルゴン酸、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、イソステアリン酸、ヒドロキシステアリン酸、オレイン酸、リノール酸、リシノール酸、アラキン酸、ベヘン酸、およびエルカ酸などの10〜20個の炭素原子を有する脂肪酸、ラウリル、ミリスチル、セチル、ヘキサデシル、ステアリル、イソステアリル、ヒドロキシステアリル、オレイル、リシンオレイル、ベヘニル、エルシル、および2−オクチルドデカニルアルコールなどの10〜20個の炭素原子を有する脂肪アルコール、ラノリン、ラノリン油、ラノリンロウ、ラノリンアルコール、ラノリン脂肪酸、ラノリン酸イソプロピル、エトキシル化コレステロール、プロポキシル化ラノリンアルコール、アセチル化ラノリンアルコール、リノレイン酸ラノリンアルコール、リシンオレイン酸ラノリンアルコール、リシンオレイン酸ラノリンアルコールの酢酸塩、エトキシル化アルコール−エステルの酢酸塩、ラノリンの水素添加分解、エオキシル化硬化ラノリン、および液体および半固体ラノリン吸収基剤などラノリンおよびラノリン誘導体、エチレングリコールモノおよびジ−脂肪酸エステル、ジエチレングリコールモノ−およびジ−脂肪酸エステル、ポリエチレングリコール(200−6000)モノ−およびジ−脂肪酸エステル、プロピレングリコールモノ−およびジ−脂肪酸エステル、ポリプロピレングリコール2000モノオレイン酸、ポリプロピレングリコール2000モノステアリン酸、エトキシル化プロピレングリコールモノステアリン酸、グリセリルモノ−およびジ−脂肪酸エステル、ポリグリセロールポリ脂肪エステル、エトキシル化グリセリルモノステアリン酸、1,2−ブチレングリコールモノステアリン酸、1,2−ブチレングリコールジステアリン酸塩、ソルビタン脂肪酸エステル、およびポリオキシエチレンソルビタン脂肪酸エステルなどの多価アルコールエステル、蜜ロウ、鯨ろう、ミリスチン酸ミリスチル、ステアリン酸ステアリルなどのろうエステル、エーテルエステルの混合物を形成する各種エチレンオキシド含量のエトキシル化ソルビトールとの蜜ロウの反応生成物であるポリオキシエチレンソルビトール蜜ロウなどの蜜ロウ誘導体、カルナバおよびカンデリラロウを含むがこれらに限定されない植物ロウ、レシチンおよび誘導体などリン脂質、コレステロールおよびコレステロール脂肪酸エステルを含むがこれらに限定されないステロール、および脂肪酸アミド、エトキシル化脂肪酸アミド、および固体脂肪酸アルカノールアミドなどのアミド。
【0135】
組成物は、例えば、多価アルコール型の保湿剤を含んで成りうる。代表的な多価アルコールとしては、ポリアルキレングリコール、およびより好ましくは、プロピレングリコール、ジプロピレングリコール、ポリプロピレングリコール、ポリエチレングリコールおよびその誘導体、ソルビトール、ヒドロキシプロピルソルビトール、エリトリトール、トレイトール、ペンタエリトリトール、キリシトール、グルシトール、マンニトール、ヘキシレングリコール、ブチレングリコール(例えば、1,3ブチレングリコール)、ヘキサントリオール(例えば、1,2,6−ヘキサントリオール)、グリセロール、エトキシル化グリセロール、プロポキシル化リセロール、2−ピロリドン−5−カルボン酸ナトリウム、可溶性コラーゲン、フタル酸ジブチル、ゼラチンおよびこれらの混合物を含むアルキレンポリオールおよびその誘導体が挙げられる。
【0136】
別の任意の成分は、グアニジン;グリコール酸およびグリコール酸塩(例えば、アンモニウムおよび四級アルキルアンモニウム);乳酸および乳酸塩(例えば、アンモニウムおよび四級アルキルアンモニウム);そのさまざまな形態のいずれかにおけるアロエ(例えば、アロエゲル);糖およびでんぷん誘導体(例えば、アルコキシル化グルコース);ヒアルロン酸およびその誘導体(例えば、ヒアルロン酸ナトリウムなど塩誘導体);ラクトアミドモノエタノールアミン、アセトアミドモノエタノールアミン;尿素;パンテノール;糖;でんぷん;シリコーン液;シリコーンゴム;およびそれらの混合物である。プロポキシル化グリセロールも有用である。
【0137】
親水性および親油性のクリームが調査された。これらのクリームは、高生体適合性成分で構成されている。具体的には、これのクリーム系の脂質相は、オリーブ油、甘扁桃油、アボカド油、ホホバ油およびその他などの任意の植物性油、ワセリンまたはその他など鉱油、および/または脂肪酸のエステル、短、中、長鎖トリグリセリドなどの任意の合成油相で構成されうる。これらの系を調製するために、クリームに特定の物理化学的安定性を提供することが可能である皮膚科学的に許容される界面活性剤、例えば、レシチン、トゥイーン(Tween)80などが使用されうる。ゲル系製剤もバルプロ酸、その塩および誘導体の局所適用に可能性のある製剤として調査された。製剤に所望のレオロジー特性を提供することが可能である増粘剤を使用することによって、ゲルを調製することができる。この場合にも、ゲル系製剤の調製に使用される薬剤は、特定のヒト皮膚寛容性、すなわち、セピゲル(sepigel)、ジルゲル(zilgel)、カルボポール(carbopol)、キサンタンゴム、ゼラチン、PVP、PEOなどを有する必要がある。
【0138】
投与
特定の患者用の特定の局所用量レベルは、年齢、体重、一般的健康状態、性別、食習慣、および投薬歴、および患者の特定の疾患の重篤度、および使用される特定の化合物の活性、投与時間、排泄率、治療期間、他の薬剤、化合物、および/または組合せて使用される材料を含むさまざな因子に応じて用いることができる。活性化合物の適切な投与量、および活性化合物を含んでなる組成物が、患者によって変動することは理解されるであろう。最適な投与量の決定は、一般に本発明の治療のリスクまたは有害な副作用に対する治療的有用性のレベルの平衡を含む。
【0139】
インビボでの局所投与は、治療過程全体を通じて1回量、連続的、または断続的に達成されうる。投与の最も有効な手段および投与量を決定する方法は当業者に周知であり、治療に使用される製剤、治療の目的、治療される標的細胞、および治療される対象によって変動する。1回または多数の投与が、治療医師によって選択される用量レベルおよびパターンで実施されうる。
【0140】
一般に、適切な用量の活性化合物、すなわち、VPAまたはVPA誘導体は、少なくとも約0.1重量%、好ましくは、0.1重量%〜6重量%、より好ましくは、0.3重量%〜5重量%、より好ましくは、0.5重量%〜4重量%、かつさらにより好ましくは、1重量%〜4重量%の濃度を含んで成り、活性成分は医薬的に許容される担体に懸濁される。活性成分が塩、エステル、プロドラッグなどである場合、投与される量は親化合物に基づき計算され、こうして使用される実重量は比例して増大する。
【0141】
活性化合物、すなわち、VPAまたはVPA誘導体がレチノイドと組合せて使用される場合は、本活性化合物の適切な用量は、少なくとも0.1重量%、好ましくは、0.1重量%〜6重量%、より好ましくは、0.3重量%〜5重量%、より好ましくは、0.5重量%〜4重量%、かつさらにより好ましくは、1重量%〜4重量%の範囲であり、使用されるレチノイドについては、活性化合物の適切な用量は約0.01重量%〜約1重量%、好ましくは、0.05重量%〜0.5重量%の範囲である。
【0142】
活性化合物、すなわち、VPAまたはVPA誘導体が5−フルオロウラシルと組合せて使用される場合は、本活性化合物の適切な用量は、少なくとも0.1重量%、好ましくは、0.1重量%〜6重量%、より好ましくは、0.3重量%〜5重量%、より好ましくは、0.5重量%〜4重量%、かつさらにより好ましくは、1重量%〜4重量%の範囲であり、使用される5−フルオロウラシルについては、活性化合物の適切な用量は約0.1重量%〜約10重量%、好ましくは、1重量%〜10重量%の範囲である。
【0143】
本発明の好ましい実施態様においては、VPAは、毎日1回〜3回、蓄積総1日量0.5mg(ミリグラム)〜10mg(ミリグラム)で約1cm2(qcm)の病変当りに局所使用される。より好ましい実施態様においては、VPAは、毎日1回〜3回、蓄積総1日量1mg(ミリグラム)〜8mg(ミリグラム)で約1cm2(qcm)の病変当りに局所使用される。さらにより好ましい実施態様においては、VPAは、毎日1回〜3回、蓄積総1日量2mg(ミリグラム)〜6mg(ミリグラム)で約1cm2(qcm)の病変当りに局所使用される。実際の1日量は、治療される病変のサイズによって決まり、したがって、1日量の増大に従って約100cm2(qcm)までの病変サイズを含めて、病変サイズの増大とともに増加する。
【0144】
本発明の他の好ましい実施態様においては、VPAを含有する局所製剤は、リストアップされた成分(含量は%[w/w]で示されている)を含有する以下の3つの製剤によって構成されている。
【0145】
製剤例1:
白鉱油 20
セチルアルコール 24
セトマクロゴール1000 6
バルプロ酸 >0.1、例えば、0.1〜6
白ワセリン 100に添加
【0146】
製剤例2:
酸化亜鉛 25
でんぷん 25
バルプロ酸 >0.1、例えば、0.1〜6
白ワセリン 100に添加
【0147】
製剤例3:
白ワセリン 25
セチルアルコール 10
トゥイーン(Tween)60 5
グリセリン 10
EDTA 0.2
香水 (場合により) 2滴
バルプロ酸ナトリウム >0.1、例えば、0.1〜6
再蒸留水 100に添加
【実施例】
【0148】
実施例1
VPAまたはレチノイン酸誘導体タザロテン単独、およびVPAとタザロテンの組合せによる治療時のヒト不死化ケラチン生成細胞(HaCaT細胞)における相乗的細胞増殖阻害およびアポトーシスの誘導(図1および5)。
【0149】
方法:
MTTアッセイ:この比色アッセイでは存在する細胞の数に比例するホルマザンの形成を検出する。薬剤を含有する組織培養培地を除去し、2mg/ml MTT((3−4,5−ジメチルチアゾール−2イル)臭化2,5−ジフェニルテトラゾリウムブロミド)を含有するDME 200μlを各ウェルに添加した。96ウェルプレートを37℃、10%CO2下に30分間インキュベートした。次いで、MTTを含有する培養培地を除去し、細胞をPBSで1回洗浄した。PBSの除去後、200μL DMSOを各ウェルに添加し、プレートを回転プラットフォームシェーカー上に5分間配置した。分光光度計を使用して550nmの波長で吸光度を測定することによって結果を得た。結果は、対照を100%とするパーセンテージで示されている。
【0150】
アポトーシスアッセイ:細胞を含有するSub−G1 DNAのパーセンテージを分析することでベクトン・ディッキンソン(Beckton Dickinson)FACScaliburフローサイトメトリーによってアポトーシスを測定した。10000個のイベントが各サンプルで記録された。増幅スケールはすべてのパラメータで線形であった。光電子増倍管電圧を、FL2−H対SCC−Hグラフにおけるチャンネル300でG0/G1細胞に相当するピークDNA含量を配置するように設定した。ピークDNAよりも低い含量を示す細胞(低二倍体(hypodiploid)細胞)および高SSC−H(顆粒高凝縮細胞)をアポトーシスとみなした。実験を2通りで3回繰り返した。エラーバーは1標準偏差を示す。
【0151】
結果:
図1は、表示された化合物で3日間処理したヒト不死化ケラチン生成細胞(HaCaT細胞)で行われた増殖アッセイの結果をグラフで示す。薬剤をプレーティングと同じ日に添加した(0日目)。MTTアッセイ(細胞増殖を測定するために)を3日目に行った。薬剤のさまざまな組合せを増殖アッセイで使用した。図に示すように、単独で使用されたVPAおよびタザロテンは細胞増殖を阻害し、タザロテン+ヒストンデアセチラーゼ阻害剤VPAの組合せは、MTTアッセイによって測定されるように、両方の薬剤の単独の阻害よりも細胞増殖の阻害でより有効であった。
【0152】
同様に、VPAはHaCaT細胞におけるタザロテン誘発アポトーシスを増強する(図5)。細胞を0日目に直径35mmのウェルにプレーティングした。薬剤を1日目に添加した。アポトーシスアッセイを2日目に行った。VPAは、subG1 DNA含量の明らかな増大を誘発し、これがヒト不死化ケラチン生成細胞HaCaTのアポトーシスを誘発することを示した。図5に示されているように、タザロテン+VPAの組合せは、単独で使用された両方の薬剤で確認されたアポトーシス率よりもアポトーシスの誘発でより有効であった。したがって、これら2つの薬剤の組合せ活性は、この処理の相乗的活性によって説明されなければならない。
【0153】
実施例2
VPAまたはレチノイン酸(RA)単独、およびVPAとレチノイン酸(RA)の組合せによる処理でのヒト不死化ケラチン生成細胞(HaCaT細胞)における相乗的細胞増殖阻害およびアポトーシスの誘導(図2)。
【0154】
方法:
MTTアッセイ:詳細については実施例1を参照。
【0155】
結果:
図2に示されているように、単独で使用されたVPAおよびRAはヒトケラチン生成細胞HaCaTの増殖を阻害し、両方の薬剤を組合せて使用すると、VPAはRA誘発細胞増殖阻害をさらに増強した。細胞を0日目にウェルにプレーティングした。次いで、薬剤を指示濃度で添加した(VPA1+RA1:VPA 1mM+レチノイン酸1μM、VPA1+RA2:VPA 1mM+レチノイン酸2μM)。MTTアッセイを3日目に行った。薬剤のさまざまな組合せを増殖アッセイでも使用した。RA+ヒストンデアセチラーゼ阻害剤VPAの組合せは、MTTアッセイによって測定されるように、単独で使用された各薬剤よりも細胞増殖の阻害でより有効でった。したがって、これら2つの薬剤の組合せ活性は、この治療の相乗的活性によって説明されなければならない。
【0156】
実施例3
VPAまたは5−フルオロウラシル単独、およびVPAと5−フルオロウラシル(5−FU)の組合せによる処理でのヒト不死化ケラチン生成細胞(HaCaT細胞)における相乗的細胞増殖阻害およびアポトーシスの誘導(図3)。
【0157】
方法:
MTTアッセイ:詳細については実施例1を参照。
【0158】
結果:
単独で使用されたVPAおよび5−FUはHaCaT細胞の増殖を阻害し、VPAは、両方の薬剤を組合せて使用すると、これらの細胞における5−FU誘発増殖阻害を増強した(図3)。薬剤をプレーティングと同じ日に添加した(0日目)。MTTアッセイ(細胞増殖を測定するために)を3日目に行った。5μMまたは10μM 5−FUおよび1mM VPAを使用した。5−FU+HDAC阻害剤VPAの組合せは、MTTアッセイによって測定されるように、単独で使用された各薬剤よりも細胞増殖の阻害でより有効でった。したがって、これら2つの薬剤の組合せ活性は、この治療の相乗的活性によって説明されなければならない。
【0159】
実施例4
VPAは、ヒト不死化ケラチン生成細胞(HaCaT細胞)における細胞周期停止を誘発する(図4)。
【0160】
方法:
BrdU取込みアッセイ:細胞増殖をBrdUで細胞を標識化することによって検出した。指示濃度でVPAによる処理の24時間後、メーカー(ZYMED、カリフォルニア州(CA)、サンフランシスコ(San Francisco))のプロトコールに従いBrdU(1:100)で細胞をインキュベートした。細胞を1時間、培地中でインキュベートし、次いで70%エタノールで固定した。取込みBrdUを間接免疫ペルオキシダーゼ法(アマシャム(Amersham)、イリノイ州(IL)、アーリントンハイツ(Arlington Heights))。手短に言えば、培養細胞を最初に1時間、ビオチン−マウス抗BrdU抗体でインキュベートした。TTBS緩衝液(20mMトリス(Tris)、500mM NaCl、0.05%トゥイーン(Tween)−20、pH7.5)中で洗浄後、細胞をさらにビオチン化ヤギ抗マウス免疫グロブリンで約10分間インキュベートした。次いで、細胞を洗浄し、酵素(ペルオキダーゼ)複合体で10分間、室温下にインキュベートした。基質−色原体の添加によって免疫反応を示した。陽性細胞を無作為に選んだ10箇所の顕微鏡視野で計算した。
【0161】
結果:
HaCaT細胞を直径35mmのウェルに0日目にプレーティングした。VPAを指示濃度で0日目に添加した。BrdUアッセイを2日目に行った。図4に示されているように、VPAはBrdU陽性細胞の用量依存減少を引き起こし、これが細胞周期停止および/またはヒトケラチン生成細胞の細胞生存の喪失を誘発することを示した。
【0162】
実施例5
3%(w/w)で局所適用されるVPAは、時間依存様態でヒト皮膚に透過する(図6)。
【0163】
方法:
局所適用可能な水性VPA製剤の調製:セピゲル(Sepigel)(セピック(Sepic)、イタリア、ミラノ(Milan))(2%w/w)およびバルプロ酸ナトリウム(3%w/w)を適量の蒸留水中に分散させ、室温下に持続的に攪拌しながら局所バルプロ酸ナトリウム製剤を調製した。結果として生じるゲルを使用前の24時間、室温下に保存した。
【0164】
インビトロ皮膚透過アッセイ:表皮角質層(SCE)を有する細胞膜でアセンブリしたフランツ(Franz)型ガラス拡散セル(LGA、米国、カリフォルニア州(CA)、バークリー(Berkeley))を使用して分析を行った。成人ヒト皮膚サンプル(平均年齢29±5歳)を腹部減量手術から得た。皮下脂肪を切除し、皮膚サンプルを60℃下の蒸留水中に1分間、浸漬し、次いで表皮角質層(SCE)を剥離した。SCEサンプルを約25%相対湿度下に維持した乾燥機で室温下に乾燥させた。乾燥サンプルをアルミニウムホイルに包み、使用まで4℃下に保存した。乾燥SCEのサンプルをフランツ(Franz)型拡散セルに載せられる前の1時間、室温下、蒸留水中に浸漬することによって再水和した。露出皮膚表面積は0.75cm2であり、受容体容積は4.5mlであった。受入溶液、NaCl0.9%(w/w)の溶液を攪拌し、実験中に35℃下に自動温度調節した。このプロトコールで使用されるSCE膜の皮膚バリアの完全性をそのトリチウム水透過係数(Kp)を測定することによって評価した。Kp値は、1.58±0.3×10-3cmh-1であることがわかり、対応する文献において以前に報告されたものと一致した(ブロナウ(Bronaugh)ら、1986年、J Invest Dermatol 87、p.451−453)。バルプロ酸ナトリウム(バルプロエート(Valproate))を3%(w/w)で皮膚表面に局所適用した。各実験を2通りで3種類の皮膚ドナーで行った。受入溶液のサンプルを実験期間中(10時間)の異なる時点で回収し、サンプル量を同量の新しい溶液に取り替えた。全サンプルを分光蛍光検出とともにHPLCによってバルプロ酸ナトリウム含量について分析した。結果を透過総量(mg)で表した。
【0165】
バルプロ酸ナトリウムの定量:バルプロ酸ナトリウムを測定するために、4−ブロモメチル−7−メトキシクマリン(BMMC)でサンプルを誘導体化した。シクロヘキサンカルボン酸を内部標準として使用した。誘導体化は以前に報告されたように行った(ブスケ(Bousquet)ら、1991年、Pharmazie 46、p.257−258)。分析は、20μlレオダイン(Rheodyne)モデル7125注入弁(レオダイン(Rheodyne)、カリフォルニア州、コタチ(Cotati))を取付けた1050ヒューレットパッカード(Hewlett Packard)液体クロマトグラファーから構成されたクロマトグラフィーシステムで行われた。クロマトグラフィー分析は、ハイパーシル(Hypersil)ODS 5μmカラム(ポリコンサルト(Policonsult)、イタリア、ローマ(Rome))で行われた。移動相は、流量1ml/分でCH3CN/H2O(65/35v/v)であった。検出は310nmで行われた。
【0166】
結果:
図6は、インビトロ透過アッセイの結果を示す。成人ヒト皮膚サンプル(皮膚表面積は0.75cm2であった)を方法の部に記載したように調製した。適量のバルプロ酸ナトリウム(バルプロエート(Valproate))を3%(w/w)でフランツ(Franz)型ガラス拡散セルに配置した皮膚に局所適用した。各実験を2通りで3種類の異なる皮膚ドナーで行った。受入溶液のサンプルを実験期間中(10時間)の異なる時点で回収し、サンプル量を同量の新しい溶液に取り替えた。全サンプルを分光蛍光検出とともにHPLCによってバルプロ酸ナトリウム含量について分析した。結果を透過総量(mg)で表した。3%(w/w)で局所適用されたVPAは、時間依存様態で皮膚に透過する。したがって、VPAは、局所適用される場合には、有効にヒト皮膚に透過することが可能である。
【0167】
実施例6
VPAは、ヒト不死化ケラチン生成細胞(HaCaT細胞)におけるヒストンH4アセチル化を濃度依存様態で誘発する(図7)。
【0168】
方法:
ウェスタンブロット:HaCaT細胞を処理24時間前に6ウェル培養皿へ接種した。培養物を指示濃度のVPAおよび/またはタザロテンで24時間処理した。全細胞抽出物を12%変性ポリアクリルアミドゲル上での変性SDSゲル電気泳動のためのRIPA緩衝液+プロテアーゼ阻害剤中の細胞溶解によって調製した。抗アセチル化H4抗体(クローンT25、特許出願EP02.021984.6号を参照)を使用するウェスタンブロット分析によってアセチル化ヒストンH4を検出し、同等のローディング対照として、抗アクチン抗体(シグマ(Sigma)、カタログ番号A5441)を使用してβ−アクチンの発現を確認した。
【0169】
結果:
図7において、上パネルは、指示通り処理されたHaCaT細胞からの全細胞ライセートの代表的な抗アセチル化ヒストンH4免疫ブロットを示す。下パネルは、ローディング対照としてのレプリカゲルの抗アクチン免疫ブロットを示す。細胞を0日目に6ウェルプレートにプレーティングした。薬剤を1日目に添加し、細胞を2日目にRIPA緩衝液+プロテアーゼ阻害剤で溶解した。全細胞ライセート40μgを12.5%SDS PAGEによって分離し、指示抗体で免疫ブロット法の対象にした。図7に示されているように、VPAは用量依存様態でヒストンH4高アセチル化を誘発する。
【0170】
実施例7
3%(w/w)で局所適用されたVPAは、マウス皮膚細胞(図8および9)およびヒト基底細胞癌(BCC)細胞(図10)の核におけるヒストンH4のアセチル化を誘発する。
【0171】
方法:
免疫組織化学:3%(w/w)で局所適用されるVPA(バルプロエート(Valproate))(製剤の詳細については実施例5を参照)またはプラセボ製剤を無胸腺ヌードマウスの皮膚に適用した。同様に、インフォームドコンセントを与えた後、3%(w/w)で適用されるVPA、またはプラセボ製剤を、外科的切除の16−20時間前に患者のBCC病変に局所適用した。3μmの凍結皮膚切片を室温下に10分間、アセトン中に固定した。抗アセチル化H4抗体(クローンT25、上記参照)での染色前に、ヤギ抗マウス免疫グロブリン(カペル(Cappel))(50mMトリス(Tris)緩衝食塩水、TBS中1/10000希釈)由来のF(ab’)2断片による30分間のプレインキュベーションによって組織をブロックした。TBSによる10分間の洗浄後、室温下に1時間、抗アセチル化H4抗体(クローンT25)(TBS中1/4000希釈)で切片をインキュベートした。次いで、試料をウサギ抗マウス血清でインキュベートし、APAAP(アルカリホスファターゼ抗アルカリホスファターゼ)免疫複合体で上記の通り処理した。陽性染色を、基質としてナフトール−AS−BIリン酸塩、かつ色原体としてニューフクシン(New Fuchsin)(ダコ(Dako))で赤色に発色させた。光学顕微鏡装置に接続したデジタルカメラで写真撮影した。
【0172】
結果:
皮膚生検を切除直後に液体窒素中で凍結させた。次いで、免疫組織化学を方法で記載した通り3μmの切片で行った。プラセボまたはVPAで局所治療した皮膚の生検を並行して処理し、同じ光度下にデジタルカメラに接続した光学顕微鏡で写真撮影した。
【0173】
図8および9に明らかに示されているように、局所VPA治療は、無胸腺ヌードマウスの皮膚細胞において、かつ―最も重要なことには―患者のBCC細胞および正常な皮膚細胞両者にヒストンH4アセチル化の明らかな増大を誘発した(図10)。これらのデータは、皮膚に局所適用されるVPAが皮膚細胞の高アセチル化を有効に誘発しうることを示す。
【0174】
実施例8
VPAは、0.1%(w/w)で適用されるタザロテンおよび3%(w/w)で適用されるVPAによる局所治療の前後6週間の代表的ば患者3例の写真に示されるように、表在性BCCの治療におけるタザロテンの効果を増強する(図11)。
【0175】
方法:
そのインフォームドコンセントを与えた後、平均年齢68歳の患者10例(男性6例、女性4例)を登録し、0.1%(w/w)のタザロテンおよび3%(w/w)で局所適用されるVPAで8週間1日1回、局所治療した。10個の病変、すなわち表在性BCC8個および色素性BCC2個を治療した。これらの病変のうち4個はすでにタザロテン単独で少なくとも16−20週間、前治療されており、この治療に対して完全に耐性であるように思われ、すなわち、タザロテン治療下に腫瘍直径の削減が確認されなかった。VPAをタザロテン適用の15−30分前に1日1回、局所適用した。病変を臨床的に評価し、2週間おきに写真を撮った。
【0176】
結果:
局所適用VPAおよびタザロテンによるBCC患者の治療は、6〜8週間の治療において全病変の50%を上回る後退(直径削減によって測定)をもたらした。これらの結果は、以前に未治療だった患者、および以前にタザロテンのみに暴露された患者の両方で確認された。興味深いことに、単独使用されたタザロテンは、BCCの数の後退(約50%)を誘発することがわかっているが、後退は通常ゆっくり(12−24週間)であり、一部の望ましくない副作用(疼痛や掻痒など)を伴う。しかし、VPAおよびタザロテンを組合せて使用する局所治療は明らかにタザロテンに対する反応を加速し、タザロテンによって引き起こされる望ましくない副作用の持続時間をも削減する。
【0177】
これらの結果により、VPAが他の化合物(タザロテンなど)の腫瘍細胞、特に皮膚の腫瘍の細胞に対する効果を増強しうるとともに―タザロテン耐性BCCの場合―VPAが腫瘍の耐性形態を感受性形態に変換しうることを示すことが確認される。全体的に、これらの結果により、皮膚過剰増殖性疾患の局所治療におけるVPAの有効性が確認される。
【0178】
実施例9
製剤例(軟膏)
白鉱油 20%
セチルアルコール 24%
セトマクロゴール1000 6%
バルプロ酸 3%
白ワセリン 100%に添加
【0179】
実施例10
製剤例 (ペースト)
酸化亜鉛 25%
でんぷん 25%
バルプロ酸 3%
白ワセリン 100%に添加
【0180】
実施例11
製剤例 (クリーム)
白ワセリン 25%
セチルアルコール 10%
トゥイーン(Tween)60 5%
グリセリン 10%
EDTA 0.2%
バルプロ酸ナトリウム 3%
再蒸留水 100%に添加
【0181】
実施例12
炎症性サイトカインなど免疫学的に関連するタンパク質の発現の調節。異なるHDAC阻害剤によるヒト不死化ケラチン生成細胞および末梢血リンパ球の処理は、結果として炎症性サイトカインの減少をもたらす(図12〜16)。
【0182】
方法:
ヒト不死化ケラチン生成細胞からの全RNAの単離
ヒト不死化ケラチン生成細胞(HaCaT細胞)を250万細胞/mlの密度で75cm2フラスコの中へ接種した。細胞を未処理で放置するか、または200nMトリコスタチンA(TSA)または5mMバルプロ酸(VPA)で4時間37℃下にプレインキュベートした後、リポ多糖類(LPS)(100ng/ml)で刺激した。37℃下に24時間後、細胞を溶解し、キアゲン(Qiagen)のRNeasyミニキットを使用して全RNAを単離した。
【0183】
末梢血単核細胞の単離および処理
単球およびマクロファージ枯渇末梢血単核細胞をFicoll−Hypaqueを使用する分離によって同意した成人から得た。末梢血単核細胞(PBMC)画分を洗浄し、9cmのペトリ皿に接種した。37℃下に2時間のインキュベーション後に単球、マクロファージ、およびB細胞の大部分を除去し、非接着細胞を回収し、175cm2のフラスコで2日間培養した。細胞を採取し、300万細胞/mlに調整した。500μlのアリコートを24ウェル平底プレートの各ウェルに移した。末梢血リンパ球(PBL)を指示通りさまざまな濃度のHDAC阻害剤で処理した。37℃下の2時間のインキュベーション後、細胞をホルボール12−ミリスチン酸13−アセテート(PMA)/イオノマイシン(Ion)で刺激し、または10μg/ml抗CD3mAb(OKT3)および2.5μg/ml抗CD28mAbとのT細胞受容体(TCR/CD3)複合体によって活性化した。37℃下に24時間後、上清を除去し、サイトカインアッセイ用に凍結した。細胞ペレットを溶解し、キアゲン(Qiagen)のRNeasyミニキットを使用して全RNAを単離した。
【0184】
RT−PCRおよび半定量PCR
全RNAの1マイクログラムを逆転写酵素およびオリゴ−dTプライマー(インビトロジェン(Invitrogen))使用する標準方法によってcDNAに転写した。半定量PCRのために、cDNA 2μlを特異的プライマーを使用するPCRによって増幅させた。PCR用のプライマーはMWGによって合成したが、それは以下の通りである。
【0185】
GAPDH: 5’−GGTGAAGGTCGGAGTCAACG−3’(配列番号:1)および
5’−CAAAGTTGTCATGGATGACC−3’(配列番号:2)、
IL−2: 5’−ATGTACAGGATGCAACTCCT−3’(配列番号:3)および
5’−TCAAGTTAGTGTTGAGATGA−3’(配列番号:4)、
IL−4: 5’−ATGGGTCTCACCTCCCAACT−3’(配列番号:5)および
5’−TCAGCTCGAACACTTTGAAT−3’(配列番号:6)、
IL−5: 5’−ATGAGGATGCTTCTGCATTTGAG−3’(配列番号:7)および
5’−TCCACTCGGTGTTCATTACACC−3’(配列番号:8)、
IL−6: 5’−ATGAACTCCTTCTCCACAAGCGCC−3’(配列番号:9)および
5’−CTACATTTGCCGAAGAGCCCTCAG−3’(配列番号:10)、
IL−8: 5’−ATGACTTCCAAGCTGGCCGTGGC−3’(配列番号:11)および
5’−TTATGAATTCTCAGCCCTCTTC−3’(配列番号:12)、
IL−10: 5’−TTGCCTGGTCCTCCTGACTG−3’(配列番号:13)および
5’−GATGTCTGGGTCTTGGTTCT−3’(配列番号:14)、
IL−12: 5’−ATGTGTCACCAGCAGTTGGTCATC−3’(配列番号:15)および
5’−CTATAGTAGCGGTCCTGGGC−3’(配列番号:16)、
TNF−α: 5’−ATGAGCACTGAAAGCATGATCCGG−3’(配列番号:17)および
5’−TCACAGGGCAATGATCCCAAAG−3’(配列番号:18)、
IFN−γ: 5’−ATGAAATATACAAGTTATATCTTGGCTTT−3’(配列番号:19)および
5’−TTACTGGGATGCTCTTCGAC−3’(配列番号:20)。
【0186】
IL−2およびTNF−αELISA
ELISAを行うために、処理および未処理PBLの上清を回収し、デュオセット(Duo Set)ELISA開発システム(R&Dシステムズ(R&D Systems))を使用し、メーカーによって記載されているようにIL−2およびTNF−αを測定した。
【0187】
ウェスタンブロット
全細胞抽出物をプロテアーゼ阻害剤を含む溶解緩衝液中で細胞溶解によって調製した。ライセートをSDSゲル電気泳動によって分離し、PVDF膜上に移した。アセチル化ヒストンH3およびH4を抗アセチル化H3抗体(アップステート(Upstate)、#06−942)、抗アセチル化H4抗体(クローンT25、特許出願EP02.021984.6号)、および抗−β−アクチン抗体を使用するウェスタンブロット分析によって検出した。同等のローディング用の対照としてβ−アクチン抗体を使用した。
【0188】
結果:
TSAおよび他のHDAC阻害剤による細胞の処理は、ヒストン高アセチル化および転写の調節をもたらす。したがって、本発明者らは、半定量RT−PCR分析によってサイトカインmRNAの発現レベル、およびELISAによってサイトカイン分泌に対するTSAおよび他のHDACの効果を調査した。
【0189】
ヒト不死化ケラチン生成細胞(HaCaT細胞)を、それぞれ、TSAまたはVPAの非存在下または存在下に24時間培養した。HDAC阻害剤で4時間のプレインキュベーション後、LPSを使用してサイトカイン産生を誘発した。サイトカインmRNA発現のレベルは半定量RT−PCRによって示されている(図12)。RT−PCR産物のアガロースゲル電気泳動は、未処理、ただし刺激対照と比べ、TSAおよびVPA処理細胞におけるTNF−αおよびIL−6mRNAのレベルにおける顕著な減少を示した(図12A)。これらの条件下、内部制御としてのGAPDH mRNAは影響を受けないままであった。ただし、LPS刺激によるIFN−γの誘導は適度に過ぎず、IFN−γのmRNA転写は実質的にTSAへの暴露によって影響されなかったが、VPAによって顕著に減少した。
【0190】
同様の結果は、末梢血リンパ球(PBL)を使用して得られた(図12B)。単離PBLをTSAおよびVPAで2時間プレインキュベートした後、37℃下に24時間PMA/Ionで刺激した。図12Bは、IL−4およびIL−6mRNA転写に対するTSAおよびVPAの効果を示す。トリコスタチンA(TSA)およびVPAは、IL−4のPMA/Ion介在刺激を顕著に減少させた。TSA処理後のIL−6mRNAでは適度の効果のみが確認されたが、VPAはIL−6mRNAを減少させ、刺激されていないサンプルで確認されるレベルに戻した。
【0191】
さらに、 図13AおよびBは、スベロイルアニリドヒドロキサム酸(SAHA)、G2M−701、G2M−702、およびG2M−707など他のHDAC阻害剤がサイトカイン発現を調節しうることを示す。
【0192】
図13に示されているように、さまざまなHDAC阻害剤は、IL−4およびIL−6mRNA転写の発現を減少させたが、GAPDHmRNA発現を修飾することはなかった。これらの条件下、IL−8mRNAは安定したままであり、PMA/IonによるT細胞活性化がこの遺伝子の発現を修飾することがないことを示した。
【0193】
相対的に、IL−2およびIFN−γ転写レベルに対するHDAC阻害剤の効果は、図14に示されているように、T細胞がCD3およびCD28抗体を使用してT細胞受容体複合体によって活性化された場合により顕著であった。PBLは、HDAC阻害剤TSA、SAHA、VPA、G2M−701、または抗炎症性ステロイドデキサメタゾン(Dex)でプレインキュベートされ、VPAおよびG2M−701は2つの異なる濃度で使用された。細胞は、24時間CD3およびCD28抗体を使用してT細胞受容体複合体(TCR/CD3)によって活性化された。図14AおよびBに示されているように、一部のサイトカインのmRNAは、わずかな相違で使用されたすべてのHDAC阻害剤によって顕著に減少した。図14Cは、アセチル化ヒストンH3およびアセチル化H4ならびに同等のローディングの対照としてのβ−アクチンに対する抗体を使用するウェスタンブロット分析を示し、HDAC阻害剤によるヒストン高アセチル化の有効な誘導を示した。
【0194】
半定量PCRの実験と一致する同様の結果が、図15に示されているようにELISAによるPBL培養上清における分泌IL−2およびTNF−αタンパク質レベルを分析することによって得られた。用量反応分析を行うために、VPA、G2M−701、G2M−702、およびG2M−707の濃度を上昇させてPBLを2時間、処理した後、CD3およびCD28mAbで24時間、37℃下に活性化した。上清を回収し、IL−2およびTNF−α分泌をELISAによって定量した。VPA、G2M−701、G2M−702、およびG2M−707によるPBLの治療は、結果として、IL−2およびTNF−α分泌の用量依存阻害をもたらした。阻害剤VPAの0.5mMおよび1mMが適度の効果のみを有したが、5mMはIL−2の分泌を顕著に減少した。
【0195】
これは、図16に示されているように、すでにVPAの1mMがIL−2およびTNF−α分泌における大幅な減少を示した、他の実験においてより顕著であった。IL−2およびTNF−α発現の阻害は、他のHDAC阻害剤でより有効であり、6μMのG2M−701、3μMのG2M−702、および1μMのG2M−707で最大であった(図15)。
【0196】
総合すれば、これらの結果は、VPA、G2M−701、G2M−702、およびG2M−707などHDAC阻害剤が、ヒトTリンパ球およびヒトケラチン生成細胞におけるサイトカイン発現のPMA/Ion(図12、13、および16)およびTCR/CD3(図14、15、および16)介在誘導を阻害することを示した。
【0197】
したがって、HDAC阻害剤は、細胞活性化に対する反応におけるサイトカイン発現を修飾する可能性を有する。それらは、免疫学的に重要な炎症性サイトカイン産生を無効にする一部のサイトカイン転写の発現をブロックすることができる。HDAC阻害剤によるサイトカイン分泌の劇的なダウンレギュレーションは、治療薬としてのその可能な使用を裏づける。
【0198】
実施例13
患者におけるHDAC阻害剤を使用する臨床治療データ
ヒストンデアセチラーゼクラスI酵素の選択的阻害剤として作用するVPAは、患者の細胞系および末梢血細胞におけるヒストン高アセチル化を誘発する。第I/II相臨床試験の範囲で静脈内VPAで治療した進行性悪性疾患を示す患者2例(患者#1および患者#2)から血液サンプルを採取した(図17、18、および19)。
【0199】
方法:
ウェスタンブロット
VPAで治療した患者からの末梢血細胞を、VPA治療の開始前、および開始後6時間、24時間、および48時間で得た(治療スケジュール、図17を参照)。全細胞抽出物をプロテアーゼ阻害剤を含むRIPA緩衝液中で細胞溶解によって調製した。ライセートをSDSゲル電気泳動によって分離し、PVDF膜上に移した。アセチル化ヒストンH3およびH4およびマーカー遺伝子HDAC−2を、抗アセチル化H3抗体(アップステート(Upstate)、#06−942)、抗アセチル化H4抗体(クローンT25、特許出願EP02.021984.6号)、および抗HDAC−2抗体(SCBT、SC−7899)を使用するウェスタンブロット分析によって検出した。同等のローディング対照PVDF膜をクーマシーで染色した(図18)。
【0200】
ELISA
VPA治療の開始前、開始後6時間、24時間、および48時間にVPAで治療した患者からの末梢血細胞を、100万細胞/mlの密度で24ウェル平底プレートへ接種した。細胞を刺激せずに放置し、またはCD3およびCD28抗体で刺激した。37℃下に24時間後、上清を回収し、IL−2およびTNF−αをELISA(R&Dシステムズ(R&D Systems))によって定量した(図19AおよびB)。
【0201】
RT−PCR
未刺激およびCD3/CD28刺激細胞からの全RNAをRNeasyミニキット(キアゲン(Qiagen))を使用して単離した。全RNAの1マイクログラムを逆転写酵素およびオリゴ−dTプライマー(インビトロジェン(Invitrogen))使用する標準方法によってcDNAに転写した。半定量PCRのために、cDNA 2μlを特異的プライマーを使用するPCRによって増幅させた(図19C)。
【0202】
結果:
末梢血細胞ライセートによるウェスタンブロット分析(図18)は、ヒストンH3およびH4高アセチル化、および治療血漿濃度を超える血清レベルでのマーカータンパク質HDAC−2のダウンレギュレーションの検出を示す。HDAC−2のヒストン高アセチル化およびダウンレギュレーションの誘導は、明らかにVPA治療の有効性を示したが、これはVPAが末梢血細胞におけるヒストン高アセチル化および標的遺伝子HDAC−2のレギュレーションを誘発する有効な治療血清濃度に達する患者において使用されうることを示す。また、本発明者らは、VPAが、ELISAによって分析されるように(図18A、およびB)培養上清におけるIL−2およびTNF−αなど炎症性サイトカインの発現を調節する証拠を示すが、これはCD3/CD28刺激細胞におけるIL−2およびTNF−α mRNA転写の減少と一致する(図18c)。さらに、これはVPA治療の24時間で開始するIL−4およびIFN−γのサイトカインmRNA発現を顕著に削減した。
【0203】
総合すれば、これらのデータは、VPAが、図17に示されている治療スケジュールによるHDAC阻害剤VPAで治療した患者におけるIL−2、TNF−α、IL−4、およびIFN−γなど免疫学的関連性遺伝子を有効に調節しうることを示す。
【0204】
したがって、免疫調節化合物として作用するVPAおよび他のHDAC阻害剤のこの新しい可能性は、病理学的に過剰活性の免疫細胞に結合される疾患の治療用の抗炎症薬としてこれらの化合物を使用する本発明を裏づける。
【図面の簡単な説明】
【0205】
【図1】VPAは、タザロテン(Taz)誘発細胞増殖阻害を増強する。 ヒト不死化ケラチン生成細胞(HaCaT細胞)で行われた増殖アッセイの結果を示し、単独で使用されたVPA、および単独で使用されたレチノイン酸誘導体タザロテンがHaCaT細胞増殖を阻害し、ヒストンデアセチラーゼ阻害剤VPAと組合せたタザロテンの使用は、単独で使用された各薬剤よりも細胞増殖の阻害でより有効であることを示す(NT:未処理、Taz:タザロテン、VPA1+Taz1:VPA 1mM+タザロテン 1μM、VPA1+Taz2:VPA 1mM+タザロテン 2μM)。
【図2】VPAは、レチノイン酸(RA)誘発細胞増殖阻害を増強する。 単独で使用されたVPAおよびRAがヒト不死化ケラチン生成細胞(HaCaT細胞)の増殖を阻害し、VPAがこれらの細胞において両方の薬剤が組合せて使用されるとレチノイン酸誘発細胞増殖阻害を増強することを示す(NT:未処理、RA:レチノイン酸、VPA1+RA1:VPA 1mM+レチノイン酸 1μM、VPA1+RA2:VPA 1mM+レチノイン酸 2μM)。
【図3】VPAは、5−フルオロウラシル(5−FU)誘発細胞増殖阻害を増強する。 単独のVPAおよび5−FUはHaCaT細胞の増殖を阻害し、5−FU(指示濃度で使用)+VPAの組合せは、単独で使用された各薬剤よりもHaCaT細胞増殖の阻害でより有効であることを示す(NT:未処理、5−FU:5−フルオロウラシル、VPAは1mMの濃度で5−FUと組合せて使用される)。
【図4】VPAは、ヒトケラチン生成細胞の細胞周期停止を誘発する。 VPAだけがヒト不死化ケラチン生成細胞(HaCaT細胞)における細胞周期停止を誘発することを示す。
【図5】VPAは、タザロテン誘発アポトーシスを増強する。 単独で使用されたタザロテンおよびVPAがHaCaT細胞のアポトーシスを誘発し、タザロテン+VPAの組合せが、単独で使用された各薬剤よりもこれらの細胞におけるアポトーシスの誘発でより有効であることを示す(NT:未処理、Taz:タザロテン、VPA1+Taz1:VPA 1mM+タザロテン 1μM、VPA1+Taz2:VPA 1mM+タザロテン 2μM)。
【図6】3%(w/w)で局所適用されるVPAは、ヒト皮膚に透過する。 3%(w/w)で局所適用されたVPAによるヒト皮膚の透過の時間依存様態を示す。
【図7】VPAは、ケラチン生成細胞におけるヒストンH4アセチル化を誘発する。 用量依存様態でHaCaT細胞におけるVPA誘発ヒストンH4アセチル化を示す。
【図8】3%(w/w)で局所適用されるVPAは、マウスにおける皮膚細胞アセチル化を誘発する。 3%(w/w)で局所適用されたVPAで処理されたマウスの皮膚細胞におけるヒストンH4アセチル化を示す。
【図9】3%(w/w)で局所適用されるVPAは、マウスにおける皮膚細胞アセチル化を誘発する。 図8における表示と比べて高い倍率における3%(w/w)でのVPAの局所適用により誘発されたマウスの皮膚細胞におけるヒストンH4アセチル化を示す。
【図10】3%(w/w)で局所適用されるVPAは、BCC核のアセチル化を誘発する。 3%(w/w)でのVPAの局所適用後のヒト基底細胞癌(BCC)におけるヒストンH4のアセチル化の誘導を示す。
【図11】3%(w/w)で局所適用されるVPAは、表在性BCCの治療におけるタザロテンの効果を増強する。 治療前、および0.1%(w/w)で適用されたタザロテンおよび3%(w/w)で適用されたVPAによる併用局所治療の6週間後の3人の代表的な患者の例を示す。
【図12】ヒトケラチン生成細胞および末梢血リンパ球におけるTSAおよびVPAによる炎症性サイトカインの調節。 LPSまたはPMA/Ionによる刺激後のHDAC阻害剤による炎症性サイトカインなど免疫学的に関連する遺伝子のmRNAの発現調節を示す(実施例12)。
【図13】異なるHDAC阻害剤による炎症性サイトカインの調節。 PMA/Ionによる刺激後のHDAC阻害剤による炎症性サイトカインなど免疫学的に関連する遺伝子のmRNAの発現調節を示す(実施例12)。
【図14】末梢血リンパ球におけるHDAC阻害剤による炎症性サイトカインの調節。 CD3/CD28刺激後のHDAC阻害剤による炎症性サイトカインなど免疫学的に関連する遺伝子のmRNAの発現調節を示す(実施例12)。
【図15】末梢血リンパ球におけるHDAC阻害剤によるIL−2およびTNF−α発現の調節。 CD3/CD28刺激後のHDAC阻害剤による炎症性サイトカインなど免疫学的に関連する遺伝子のタンパク質の発現調節を示す(実施例12)。
【図16】PMA/IONおよびCD3/CD28mABで刺激した末梢血リンパ球におけるHDAC阻害剤によるIL−2およびTNF−α発現の調節。 HDAC阻害剤による炎症性サイトカインなど免疫学的に関連性の遺伝子のタンパク質発現調節を示す(実施例12)。
【図17】第I/II相試験からの患者のVPA治療スケジュール。 癌患者におけるHDAC阻害剤VPAを用いた臨床治療スケジュール(実施例13)を示す。
【図18】VPAは、第I/II相試験からの患者の末梢血におけるヒストン高アセチル化およびマーカー遺伝子の制御を誘発する。 図17に示されている治療スケジュールによるHDAC阻害剤VPAにより治療された患者の末梢血におけるHDAC−2タンパク質のヒストンアセチル化およびダウンレギュレーションの有効な誘導を示す(実施例13)。
【図19】第I/II相試験における患者からのVPAによる炎症性サイトカインの調節。 CD3/CD28刺激後の図17に示されている治療スケジュールによる患者におけるHDAC阻害剤VPAを使用することによって炎症性サイトカインなど免疫学的に関連する遺伝子の有効なmRNAおよびタンパク質の発現調節を示す(実施例13)。
【技術分野】
【0001】
本発明は、ヒト癌、例えば基底細胞癌、扁平上皮細胞癌、角化棘細胞腫、ボーエン病、皮膚T細胞リンパ腫などの局所治療用とともに、前悪性病変(光線性角化症など)、および皮膚および/または粘膜の炎症(乾癬、魚鱗癬、座瘡など)の局所治療用の単独またはレチノイドもしくは核内受容体リガンド、または化学療法剤(例えば、5−フルオロウラシル)の局所適用製剤と組合せて使用されうるバルプロ酸またはその誘導体を含有する局所適用製剤に関する。本発明は、紫外線からの保護用、および日焼けの治療用の本局所適用製剤の使用にも関する。また、本製剤は、ヒストンの低アセチル化と関係があり、または高アセチル化の誘導が、例えば、形質転換皮膚および粘膜細胞における分化および/またはアポトーシスの誘導によって有利な効果を有する疾患において使用されうる。さらに、本製剤は、皮膚および/または粘膜の他の増殖性疾患の治療、および皮膚および/または粘膜組織における異常な遺伝子発現と関係がある状態の治療または予防に有用である。本発明は、上記のヒト疾患の局所治療用に臨床的に使用される薬品の製造を含む。
【背景技術】
【0002】
クロマチン制御と癌
クロマチンの局所再構成は、遺伝子の転写活性化における重要なステップである。DNAのヌクレオソームパッケージングにおける動的変化が生じ、転写タンパク質をDNA鋳型と接触させる必要がある。クロマチン再構成および遺伝子転写に影響を及ぼす最も重要な機序の1つは、クロマチン構造におけるアセチル化およびその後の変化によるヒストンおよび他の細胞タンパク質の翻訳後修飾である(デービー(Davie)、1998年、Curr Opin Genet Dev 8、p.173−178、コウザライズ(Kouzarides)、1999年、Curr Opin Genet Dev 9、p.40−48、ストラール(Strahl)とアリス(Allis)、2000年、Nature 403、p.41−44)。ヒストン高アセチル化の場合、DNAに対する静電引力の変化および疎水性アセチル基によって導入される立体障害が、DNAとのヒストンの相互作用の不安定化をもたらす。結果として、ヒストンのアセチル化がヌクレオソームを崩壊させ、DNAが転写機構にアクセスしやすくなることを可能にする。アセチル基の除去は、ヒストンがDNAおよび隣接したヌクレオソームとよりしっかりと結合し、したがって転写抑制されたクロマチン構造を維持することを可能にする。アセチル化は、ヒストンアセチルトランスフェラーゼ(HAT)活性を有する一連の酵素によって介在される。逆に言えば、アセチル基が特異的ヒストンデアセチラーゼ(HDAC)酵素によって除去される。これらの機序の崩壊は転写ミスレギュレーションを生じさせ、腫瘍化および腫瘍進行の原因となりうる。
【0003】
また、転写因子など他の因子は、そのアセチル化状態によって活性および安定性を変化させる。例えば、急性前骨髄球白血病(APL)と関係がある融合タンパク質であるPML−RARは、p53の脱アセチル化および分解を介在することによりp53を阻害し、したがってAPLブラストがp53依存癌監視経路を回避することを可能にする。造血前駆細胞におけるPML−RARの発現は結果として、p53介在転写活性化の抑制、および遺伝毒性ストレス(X線、酸化的ストレス)によって誘因されるp53依存アポトーシスからの保護をもたらす。しかし、p53の機能は、p53阻害の基礎を成す機序としてPML−RARによるp53へのHDACの積極的関与にかかわるHDAC阻害剤の存在下に再インストールされる(インシンガ(Insinga)ら、2003年、投稿中)。したがって、タンパク質のアセチル化は、p53のアセチル化などヒストンと異なり、HDAC阻害剤の抗腫瘍活性における重要な役割を果たす。
【0004】
核内受容体とヒストンデアセチラーゼ
核内ホルモン受容体は、遺伝子発現の陽性および陰性対照により発生および恒常性を制御するリガンド依存転写因子である。これらの制御過程における欠陥が、多くの疾患の原因の基礎を成し、癌の発生における重要な役割を果たす。T3R、RAR、およびPPARを含む多くの核内受容体は、リガンドの非存在下にN−CoRおよびSMRTなどコリプレッサーと相互作用し、それによって転写を阻害しうる。さらに、N−CoRは、アンタゴニストと結合したプロゲステロンおよびエストロゲン受容体と相互作用することも報告されている。最も興味深いことに、N−CoRおよびSMRTは、mSin3タンパク質およびヒストンデアセチラーゼをも含有する大きなタンパク質複合体に存在することが示されている(パジン(Pazin)とカドナガ(Kadonaga)、1997年、Cell 89、p.325−328)。したがって、核内受容体の抑制から活性化へのリガンド誘発スイッチは、コリプレッサーおよび補助活性化因子の複合体の拮抗酵素活性への変換を示す。
【0005】
核内受容体による遺伝子制御
HDAC活性を含有するかかるコリプレッサー複合体は、核内受容体による抑制を介在するだけではなく、Mad−1、BCL−6、およびETOを含む別の転写因子と相互作用もする。これらのタンパク質の多くは、細胞増殖および分化の疾患において重要な役割を果たす(パジン(Pazin)とカドナガ(Kadonaga)、1997年、Cell 89、p.325−328、ヒューン(Huynh)とバードウェル(Bardwell)、1998年、Oncogene 17、p.2473−2484、ワング(Wang),J.ら、1998年、Proc Natl Acad Sci USA 95、p.10860−10865)。例えば、T3Rは最初は、野生型受容体とは対照的にリガンドに結合することはなく、転写の構成的コリプレッサーとして機能するそのウイルス癌遺伝子v−erbAとの相同性に基づき同定された。さらに、RARにおける突然変異は、多くのヒト癌、特に急性前骨髄球性白血病(APL)および肝細胞癌と関係がある。APL患者においては、染色体転座由来のRAR融合タンパク質が、前骨髄球白血病タンパク質(PML)または前骨髄球亜鉛フィンガータンパク質(PLZF)のいずれかを伴う。両方の融合タンパク質はコリプレッサー複合体の成分と相互作用しうるが、レチノイン酸の添加によりPML−RARからコリプレッサー複合体が追放されるの対して、PLZF−RARは構成的に相互作用する。これらの所見は、PML−RARAPL患者がレチノイン酸治療後に完全寛解を達成するのに対して、PLZF−RAR APL患者がきわめて不十分に反応する理由の説明を提供する(グリニャーニ(Grignani)ら、1998年、Nature 391、p.815−818、グイデ(Guidez)ら、1998年、Blood 91、p.2634−2642、ホー(He)ら、1998年、Nat Genet 18、p.126−135、リン(Lin)ら、1998年、Nature 391、p.811−814)。
【0006】
最近、レチノイン酸による治療後に多重再発を経験したPML−RAR患者が最近、HDAC阻害剤のフェニルブチレートで治療され、結果として白血病の完全寛解がもたらされている(ワレル(Warrell)ら、1998年、J.Natl.Cancer Inst.90、p.1621−1625)。
【0007】
HDAC阻害剤による抗癌治療
追加の臨床試験では最近、HDAC阻害の原理を用いた癌患者の全身臨床治療の活用が開始されている。今のところ、単剤療法としての密接に関連した酪酸誘導体ピバネックス(Pivanex)(チタン製薬(Titan Pharmaceutical))による第II相臨床試験が完了し、第III/IV期の非小細胞肺癌における活性が明らかにされている(キール(keer)ら、2002年、ASCO、要約番号1253)。多くのHDAC阻害剤が同定されており、NVP−LAQ824(ノバルティス(Novartis)およびSAHA(アトンファルマ(Aton Pharma)社)が第II相臨床試験されたヒドロキサム酸の構造的クラスのメンバーである(マークス(Marks)ら、2001年、Nature Reviews Cancer 1、p.194−202)。他のクラスは、T細胞リンパ腫の治療用に第II相臨床試験で有効に使用されたデプシペプチド(FR901228号−藤沢(Fujisawa))など環状テトラペプチドを含む(ピエカルツ(Piekarz)ら、2001年、Blood 98、p.2865−2868)。さらに、ベンズアミドのクラスに関連した化合物であるMS−27−275(三井製薬(Mitsui Pharmaceuticals))が現在、悪性血液疾患患者を治療する第I相治験で試験されている。
【0008】
ヒストンデアセチラーゼのタンパク質ファミリー
哺乳動物のヒストンデアセチラーゼは3つのサブクラスに分類されうる(グレイ(Gray)とエクスレーム(Ekstroem)、2001年)。酵母RPD3タンパク質のホモローグであるHDAC1、2、3、および8は、クラスIを構成する。HDAC4、5、6、7、9、および10は酵母Hda1タンパク質と関係があり、クラスIIを形成する。最近、NAD依存であるデアセチラーゼの第IIIクラスを形成する酵母Sir2タンパク質の一部の哺乳動物のホモローグが同定されている。これらHDACのすべては、多くのマルチタンパク質複合体のサブユニットとして細胞に存在すると思われる。具体的には、クラスIおよびIIにHDACは、転写因子へのHDACの動員に必要な架橋因子として使用される転写補体mSin3、N−CoR、およびSMRTと相互作用することが示されている。
【0009】
バルプロ酸
バルプロ酸(VPA、2−プロピル−ペンタン酸)は、異なる分子作用機序に基づく多くの生物活性を有する。すなわち、
−VPAは、抗てんかん薬である。
−VPAは、催奇性である。妊娠中の抗てんかん薬として使用される場合、VPAは生誕児の数パーセントにおいて出生異常(神経管閉鎖障害および他の奇形)を誘発しうる。マウスにおいては、VPAは、適切に投与された場合に大半のマウス胎児において催奇性である。
−VPAは、核内ホルモン受容体(PPARδ)を活性化するいくつかの追加の転写因子はまた抑制されるが、ある因子は顕著には抑制されない(グルココルチコイド受容体、PPARα)。
−VPAは、場合により、補酵素Aにより十分に代謝されないエステルに依存した肝毒性を引き起こす。
−VPAは、HDACの阻害剤である。
【0010】
VPA誘導体の使用により、異なる活性が異なる分子作用機序によって介在されることが判明した。催奇性および抗てんかん活性は、選択的に催奇性または選択的に抗てんかんである化合物が単離されうるため、異なる作用形態をたどる(ナウ(Nau)ら、1991年、Pharmacol.Toxicol.69、p.310−321)。PPARδの活性化は、催奇性と厳密に相関し(ランペン(Lampen)ら、1999年、Toxicol.Appl.Pharmacol.、160、p.238−249)、PPARδの活性化および催奇性の両方がVPAの同じ分子活性を必要とすることが示唆された。また、F9細胞の分化は、ランペン(Lampen)ら、1999年によって示差され、かつ分化マーカーの分析によって立証されているように、PPARδの活性化および催奇性と厳密に相関する(ウェルリング(Werling)ら、2001年、Mol.Pharmacol.59、p.1269−1276)。PPARδの活性化は、VPAおよびその誘導体のHDAC阻害活性によって引き起こされることが示された(PCT/EP01/07704号、国際公開第03/024442A2号パンフレット)。さらに、既定のHDAC阻害剤であるTSAがPPARδを活性化し、かつVPAと同じ型のF9細胞分化を誘発することが示された。これらの結果から、PPARδの活性化だけではなく、F9細胞分化の誘発およびVPAまたはVPA誘導体の催奇性もHDAC阻害によって引き起こされると結論できる。
【0011】
抗てんかんおよび鎮静活性は、異なる構造活性相関をたどり、したがって、明らかにHDAC阻害と異なった一次VPA活性に依存する。肝毒性の機序の理解は不十分であり、VPA−CoAエステルの形成と関係があるかどうかは不明である。しかし、HDAC阻害は、CoAエステル形成を必要とするようには思われない。
【0012】
レチノイン酸
レチノイン酸(RA)は、遺伝子転写の調整において重要な役割を果たし、したがって、細胞分裂および分化、免疫応答および胚発生など体内の多数の機能を支配する、9−cisRA、トランスRA、全トランスRA、およびタザラトン(Tazaraton)を含むがこれらに限定されないレチノイドとして知られる化合物のクラスに属する(ギュンター(Guenther)、2003年、Am.J.Clin.Dermatol.4、p.197−202、ペリス(Peris)Kら、1999年、N Engl J Med 341、p.1767−1768)。それらはまた、癌細胞の発生および拡大を制御し、RAを含む一部は、癌細胞増殖を阻止することによって腫瘍増殖を阻害する(アルトゥッチ(Altucci)とグロンマイヤー(Gronemeyer)、2001年、Nature Reviews Cancer 1、p.181−193)。
【0013】
レチノイドの効果は2つのクラスの核内受容体、RA受容体(RAR)およびレチノイドX受容体(RXR)によって主として介在される(チャン(Zhang)とファール(Pfahl)1993年、ケストナー(Kestner)ら、1995年、マンゲルスドルフ(Mangelsdorf)とエバンス(Evans)1995年)。RARおよびRXRは、3つの異なる遺伝子(α、β、およびγ)によって暗号化される。また、多くのレチノイド受容体イソ型は、分化プロモーターの使用により生成され、多数の異なるレチノイド受容体タンパク質を生じさせる。今日まで、レチノイドの効果を介在することが知られている多数の受容体がある。9−cisRAは、RARおよびRXRの高アフィニティーリガンドであるが、トランス−RAはRARのみのリガンドである。レチノイド受容体は、多くのホルモン、ビタミン、および薬剤の生物学的効果を介在する大きなステロイド/甲状腺受容体スーパーファミリーに属する。RARおよびRXRは転写因子として作用し、活性クロマチン構造の標的遺伝子のプロモーター領域に位置したその応答エレメント(RARE)に結合することによって標的遺伝子の発現を積極的または消極的に制御し、結果として標的遺伝子の転写をもたらす。転写に対するその直接効果に加えて、リガンド化RARは、AP−1など他の転写因子の活性を調節しうる(ファール(Pfahl)1993年)。活性化レチノイド受容体はAP−1の活性を阻害し、それによってAP−1標的遺伝子の発現を制御しうる。AP−1活性の阻害は、レチノイドの抗増殖性効果と関係があり、レチノイド標的遺伝子の転写のその直接の活性化とは別であるように思われる。
【0014】
かねて医師はRA誘導体を用いていくつかの癌、特に前立腺癌および白血病を治療しており、現在ではこの薬剤で乳癌を治療する実験中である。RAによる典型的な治療では好ましい遺伝子に切り替えるためにRARを活性化することが追求される。しかし、RAに対する大きな欠点は、遺伝子を「オン」および「オフ」にするために高レベルの薬物投与を必要とすることであり、これはしばしば破壊的かつ潜在的に致死的な副作用を誘因する(アルトゥッチ(Altucci)とグロンマイヤー(Gronemeyer)、2001年、Nature Reviews Cancer 1、p.181−193)。
【0015】
RAまたはタザロテンなど他のレチノイド(ギュンター(Guenther)、2003年、Am.J.Clin.Dermatol.4、p.197−202)も軽度〜中程度の座瘡および日焼けによる損傷(光老化)皮膚の治療に局所使用されうる。レチノイドは、基底細胞癌など一部の皮膚癌を治療するために使用されている(ペリス(Peris)Kら、1999年、N Engl J Med 341、p.1767−1768)。局所投与後、RAは細胞分裂および代謝回転を増大させるように思われる。皮膚の外側部分における細胞層の数は減少する。吹出物が存在する場合、それらはより迅速に消散する。局所RAは、細いしわ、シミだらけの高色素沈着、および日光への露出過度と関係した粗さの削減において有効である。治療の結果は即効性はなく、数週間〜数か月後に明らかとなる。治療の中断は通常、結果的にRA効果の喪失をもたらす。
【0016】
基底細胞癌
基底細胞癌(BCC)は皮膚癌のもっとも一般的な形態であるだけではなく、例えば、毎年80万のアメリカ人に影響を与えるすべての癌のうち最も一般的でもある。これらの癌は、表皮(外側皮膚層)の一番下にある基底細胞に生じる。新しい症例の数は過去数十年間に毎年急に増大したが、疾患の平均発症年齢は確実に低下した。最近まで、最も多く罹患するのは高齢者、特に屋外労働していた男性であった。しかし、今日では多くの女性が過去におけるよりもBCCに罹っているが、それでも男性が依然として女性の数を大きく上回っている。
【0017】
日光への慢性的な被曝は、体の露出部分―顔、耳、首、頭皮、肩、および背中で最も頻繁に生じるほぼすべての基底細胞癌の原因である。白い肌、色の薄い髪、青色、緑色、または灰色の眼は、きわめてリスクが高い。職業が長時間屋外にいることを必要とする、または日なたで多大な余暇時間を過ごすような人は特に危険である。BCCの素因としては、紫外線、電離放射線、ヒ素、色素乾皮症および基底細胞母斑症候群などの遺伝性疾患、ワクチン接種、火傷、および瘢痕が挙げられる。
【0018】
典型的な病変は、隆起した、真珠状または半透明の境界を有する滑面結節である。病変における毛細血管拡張症が一般的な特徴である。基底細胞癌はサイズが数ミリメートル〜数センチメートルのばらつきがある。サイズは6−12か月で2倍になりうるし、または増殖がゆっくりでありうる。85パーセントが頭および首に生じ、鼻は最も一般的な部位である。患者は、手ぬぐいでの洗浄など小さな外傷による出血を報告する場合もある。この新生物は局所侵襲的であり、眼、耳、または鼻の喪失を引き起こすきわめて破壊的であり、かつ脳に侵入する場合には致死的でありうる。BCCはまれにしか再発しないが、再発する場合、リンパ管を経由して局所リンパ節に、または血行性散布によって最も一般的には肺に転移しうる。進行した皮膚癌は任意に2cmより大きい腫瘍とともに、骨筋、もしくは神経の侵襲、リンパ節転移、または美容または機能単位の除去を必要とする病変として定義されうる。
【0019】
BCCの治療は、その型、サイズ、位置、治療される数、および医師の選好または経験によって決まる。可能性としては、以下が挙げられる。すなわち、
・切除術―病変は切除され、皮膚は縫合される。
・凍結処理(凍結療法)―小さな表在性病変用に液体窒素の使用。これはBCC用の推奨治療ではない。
・掻爬術および焼灼―腫瘍の掻爬および基底の焼灼。これもBBC用の推奨治療ではない。
・放射線療法(X線治療)―進行した病変において、または手術が適切でない場合に使用されうる。
・局所化学療法―5−フルオロウラシル(5−FU)含有クリームの局所適用は、特に手術によって治療可能ではないきわめて大きな部位用に使用されうる。
【0020】
しかし、手術を嫌う、または手術が不適切な患者(例えば、ペースメーカーを付け、または出血性素因を有する虚弱で高齢の患者)、または大きな病変、多数の病変、および解剖学的に困難な部位または不十分な血管化新生皮膚を有する患者については、依然として別の有効な治療法を必要とする。
【0021】
今日、2つ以上の抗腫瘍治療用薬剤による患者の組合せ治療から成る腫瘍治療が知られている。例は、化学療法剤および/または細胞毒性試薬とともに放射線療法の併用、および腫瘍細胞特異的治療抗体の使用など、免疫学的療法と放射線療法のより最近の組合せである。しかし、個々の方法の単独よりも有効であるかかる組合せを確認するための個々の治療を互いに組合せる可能性は、広範な前臨床試験および臨床試験を必要とする。どの組合せが付加的またはさらに相乗的効果を示すか予測することは可能ではない。治療効果を増大させる目的に加えて、他の目的が、個々の成分の高用量によって引き起こされる望ましくない、または有害な副作用を減少させるために結果として生じる組合せにおける個々の成分の用量の可能な減少である。
【0022】
自己免疫および炎症におけるサイトカインの役割
サイトカインは、個々の細胞および組織の機能的活性を調節する調整剤として作用する可溶性タンパク質およびペプチドの多様なグループである。それらは、外来物質または変化した内因性物質に対する防御として炎症反応を誘発させることを目的とするものである。多くの点で、サイトカインの生物活性は、炎症、急性期反応、および自己免疫など生物学的現象を誘発する全身レベルで作用することによって特殊な腺組織における生じる典型的なホルモンのものに似ている。しかし、炎症反応の不適切な活性化は多くの一般的な疾患の基礎を成す原因であり、したがって、炎症反応は薬剤開発の重要な目標である。
【0023】
多くのサイトカインが炎症を加速させ、直接または一部の細胞型における細胞接着分子または他のサイトカインの合成を誘発するその能力により局所または全身の炎症反応を調節する。初期反応の原因となる主なサイトカインは、IL1、IL6、およびTNF−アルファである。他の炎症誘発媒体としては、LIF、IFN−ガンマ、GM−CSF、IL11、IL12、IL18、およびさまざまな他のケモカインが挙げられる。
【0024】
しかし、サイトカインの役割は、炎症過程のみに限定されるのではなく、自己免疫疾患の発症および伝播において主要な役割も有する。古典的な例が関節リウマチであり、この場合、特異的CD4+T細胞が、最も可能性があるのは未知の外生または内因性抗原に対する反応として、罹患関節において免疫応答を誘発する(オルセン(Olsen)ら、2003年、New England Journal of Medicine 350、p.2167−2179)。したがって、増強された単球、マクロファージ、および線維芽細胞は、滑液腔内の腫瘍壊死因子−α(TNF−α)およびインターロイキン−1などのサイトカインを生じる。これらのサイトカインはカスケード損傷の中核を成し、最終的にマトリクスメタロプロテイナーゼおよび破骨細胞の産生を誘因し、これが結果として軟組織および骨の不可逆性損傷をもたらす。
【0025】
活性化単球、マクロファージ、およびTリンパ球によって放出される炎症性サイトカインであるTNF−αは、関節リウマチの病因において重要である炎症反応を促進する。関節リウマチ患者は滑液中に高濃度のTNF−αを有する。TNF−αは、炎症性パンヌスおよび健康な軟骨の接合部に局在されており、高い滑液TNF−α濃度は骨の侵食と関係がある。
【0026】
当然のことながら、TNF拮抗薬は、関節リウマチ用に利用可能な特に最も有効な治療薬であるように思われる。反応は一般に迅速であり、しばしば数週間以内に発生するが、すべての患者が反応を示すわけではない。
【0027】
TNF−αに対する薬剤は、関節リウマチなど慢性自己免疫疾患の治療において有効であるだけではなく、クローン病、潰瘍性大腸炎、シェーグレン症候群、強皮症、乾癬性関節炎、強直性脊椎炎、難治性ブドウ膜炎、ベーチェット病、成人発症型スティル病、およびヴェーゲナー肉芽腫症の治療においても有効である。
【0028】
別の例が乾癬であり、この場合、T細胞介在免疫応答がケラチン生成細胞に対して行われる。これらのTリンパ球は真皮または表皮において開始抗原に直面し、1型サイトカイン(Th1)、特にインターフェロン−γ、インターロイキン2、およびTNF−αを分泌する。これらの分泌が結果としてケラチン生成細胞の増殖および成熟低下とともに関連血管変化をもたらす。インターロイキン8など他のサイトカインの分泌は、乾癬の全体像の一因となる(レブボール(Lebwohl)、2004年、The Lancet 361、p.1197−1204)。
【0029】
自己免疫疾患におけるサイトカインの因果関係の別の証拠は、さまざまな疾患の治療におけるサイトカインの使用後に得られた所見に由来する(クラウゼ(Krause)ら、2003年、The American Journal of Medicine 115、p.390−397)。興味深いことに、それらは免疫および自己免疫状態の誘因および悪化などの副作用と関係があり、これらは明白な自己免疫疾患に発展しうる。これらの自己免疫症状は、自己免疫に対する既存の傾向を有する患者においてより一般的であるように思われる。
【0030】
多発性硬化症の悪化がインターフェロン−γによる治療中に観察されている。インターフェロン−γ治療と関係した自己免疫症状の頻度は低いように思われるが、骨髄増殖性疾患用にインターフェロン−γ単独、およびインターフェロン−γと組合せて治療された患者における全身紅斑性狼瘡が報告されている。インターフェロン−γは動物モデルにおける全身紅斑性狼瘡の病因に関与している。インターフェロン−γの投与は、抗インターフェロン−γ抗体による治療によって予防される狼瘡傾向のある(NZBXNZW)F1マウスにおける糸球体腎炎への進行速度を加速する。インターフェロン−γの血清レベルの上昇が、全身紅斑性狼瘡患者において報告されている。インターフェロン−γはナチュラルキラー細胞によって生成され、II型インターフェロン受容体に結合する。これはナチュラルキラー細胞の活性化においてインターフェロン−γよりも有効ではなく、強力な抗ウイルスおよび抗腫瘍効果を有さない。しかし、インターフェロン−γは、マクロファージ活性化および腫瘍組織適合性クラスII分子の最も強力な誘発因子である。これはB細胞による免疫グロブリン分泌を刺激し、Tヘルパー1型へのT細胞の分化を促進する。
【0031】
インターロイキン2は、抗腫瘍活性を有する活性化T細胞によって分泌される。これは転移性悪性黒色腫および腎細胞癌治療において有効である。これはT細胞増殖を誘発し、B細胞増殖を増強し、ナチュラルキラー細胞および単球の活性化を促進する。インターロイキン2治療下に確認される最も一般的な自己免疫副作用は、免疫介在甲状腺疾患である。可逆性甲状腺機能不全は、インターロイキン単独またはリンホカイン活性化キラー細胞もしくはインターフェロン−γとともにインターロイキンで治療される癌患者において頻繁に生じる。転移性腎細胞癌患者におけるインターロイキン2による試験では、患者の18%(60/329)において抗甲状腺抗体が検出された。自己免疫とみなされうる他のそれほど多くない現象が、インターロイキン2治療と関係して報告されている。これらには関節リウマチ、乾癬性関節炎、強直性脊髄炎、およびライター症候群が含まれる。関節炎の誘因は、関節を浸潤し、炎症をもたらすT細胞によって認識される自己抗原の誘導によって説明されうる。インターロイキン2は、筋特異的および腫瘍抗原に対する免疫寛容の崩壊を強化し、腫瘍および筋細胞の破壊をもたらす。インターロイキン2およびリンホカイン活性化キラー細胞で治療された転移性腎細胞癌の患者が全身性硬化の急性悪化を生じた。
【0032】
インターロイキン2および可溶性インターロイキン2受容体の血清レベルは、全身性硬化症患者において上昇し、疾患期間および活性と相関する。これらの所見によりインターロイキン2治療と全身性硬化症の発症との関係が説明されうる。
【発明の開示】
【発明が解決しようとする課題】
【0033】
本発明の目的は、皮膚疾患の予防または治療用の改善された組成物を提供することである。
【課題を解決するための手段】
【0034】
VPAは、てんかんの治療に使用される薬品として開発されている。したがって、VPAは、経口または静脈内に全身使用され、薬品が血液脳関門を通り脳組織のてんかん標的部位に達し、その抗てんかん使命を果たすことを可能にする。それゆえ、VPAは、その治療効果を達成するために全身適用されるべき薬品とみなされている。
【0035】
現在、皮膚の局所に使用される治療化合物に決定的に必要とされるVPAがヒト皮膚に有効に透過することが意外にも確認された。したがって、VPAは、局所適用のこの基本的な基準を満たすことによって、皮膚の癌病変の局所治療に使用されうることが本発明者らによって仮説として提出された。
【0036】
この結果、現在、VPAは実際に、ヒト皮膚疾患、例えば、ヒト皮膚癌の局所治療に使用されうると予期しない有利な効果を有することがわかった。ここで、VPAによって使用される正確な作用形態は完全には理解されていないが、アポトーシス、成長停止、および分化誘発化合物の活性への腫瘍細胞を感作するその可能性は、かかる抗腫瘍活性の基礎でありうる。VPAのこの意外な可能性は、HDAC活性を有する一連の特異的酵素の阻害剤としてのその活性に基づくと予想される。
【0037】
したがって、本発明は、皮膚疾患の予防または治療用の局所医薬組成物に関し、
(i)VPAおよびその医薬的に許容される塩、VPAの誘導体およびその医薬的に許容される塩から成る群より選択される活性剤の少なくとも約0.1%と、
(ii)皮膚科学的に許容される担体と
を含む。
【発明を実施するための最良の形態】
【0038】
本明細書で使用される「局所適用」という語は、本発明の組成物を皮膚の表面上に適用または塗布することを意味する。
【0039】
本明細書で使用される「皮膚科学的に許容される」という語は、そのように記載される組成物またはその成分が、過度の毒性、不適合性、不安定性、アレルギー反応なしにヒト皮膚と接触して使用するために適切であることを意味する。
【0040】
別段の指示がない限り、本明細書で示されているパーセント値は重量%(w/w)である。
【0041】
本発明の組成物は、好ましくは、活性剤の約0.1%〜約25%、より好ましくは、約0.1%〜約6%、さらにより好ましくは、約0.3%〜約5%、さらにより好ましくは、約0.5%〜約4%、さらにより好ましくは、約1%〜約4%、最も好ましくは、約2%〜約4%の活性剤を含む。
【0042】
本発明の組成物は通常、活性剤、および他の任意の活性物が適切な濃度で皮膚に送達されることを可能にする、本発明の組成物が組込まれている皮膚科学的に許容される担体の約1%〜約99.9%を含む。
【0043】
好ましい実施態様においては、本発明の組成物は、25℃および大気圧で半固体である。本実施態様によれば、組成物の製品形態は、クリーム、軟膏、ゲル、またはペーストであってよい。組成物の製品形態は液分散、例えば、ローションである。
【0044】
意外にも、VPAの局所適用は、レチノイド、および5−フルオロウラシルなどの化学療法剤の治療効果を増強することがわかっている。
【0045】
本発明の他の態様においては、したがって、レチノイン酸またはその誘導体をさらに含む組成物中のレチノイン酸または誘導体の濃度は、好ましくは、組成物の約0.01%〜約1%、より好ましくは、約0.05%〜約0.5%である。レチノイドは、好ましくは、9−シスレチノイン酸、トランス−レチノイン酸、全トランスレチノイン酸、およびタザロテンから成る群より選択される。
【0046】
本発明のさらに他の態様においては、組成物は、5−フルオロウラシルなどの化学療法剤をさらに含む。化学療法剤の濃度は、好ましくは、組成物の約0.1%〜約10%、より好ましくは、約1%〜約10%である。
【0047】
好ましい実施態様においては、担体は溶液ではない。他の好ましい実施態様においては、担体はクリーム、ペースト、軟膏、ローション、またはゲルである。
【0048】
本発明の他の態様は、少なくとも約0.1%のVPAまたはその誘導体を含んでなる医薬品がそれを必要とする個人の皮膚に局所適用される、皮膚疾患の予防または治療のための医薬品を製造するためのVPA、その医薬的に許容される塩、VPAの誘導体、またはその医薬的に許容される塩の使用である。
【0049】
皮膚疾患は、好ましくは、タンパク質の高アセチル化の誘導が患者に対する有利な治療効果を有するヒト皮膚の疾患である。皮膚疾患は、皮膚腫瘍、例えば、基底細胞癌、扁平上皮細胞癌、角化棘細胞腫、ボーエン病、および皮膚T細胞リンパ腫である。
【0050】
皮膚疾患は、光線性角化症などの前腫瘍性皮膚疾患でありうる。他の実施態様においては、VPAまたはその誘導体は、皮膚および/または粘膜の炎症の治療に使用されうる。皮膚および/または粘膜の炎症の限定されない例としては、乾癬、魚鱗癬、および座瘡である。皮膚疾患は太陽で痛んだ皮膚(光老化皮膚、日焼け)でもありうる。
【0051】
本発明によるVPAまたはその誘導体の投与は、確立された抗癌治療と組合せられうる。VPAまたはその誘導体および確立された癌治療は、同時に、または連続的に(異なる時点で)適用されうる。
【0052】
別の任意の活性物質が本発明による治療において使用されうる。VPAまたはその誘導体および別の活性物質を、同時に、または連続的に(異なる時点で)投与することができる。別の活性物質としては、NVP−LAQ824(ノバルティス(Novartis))、トリコスタチンA、スベロイルアニリドヒドロキサム酸(アトン(Aton))、CBHA (アトン(ATON))、ピロキサミド(Pyroxamide)(アトン(Aton))、スクリプタイド(Scriptaid)(ジョーンズ・ホプキンス(Johns Hopkins))、CI−994 (ファイザー(Pfizer))、CG−1521 (キルカゲン(CircaGen))、クラミドシン(Chlamydocin)(ヤンセン(Janssen))、ビアリールヒドロキサム酸塩、例えば、A−161906 (アボット(Abbott))、二環式アリール−N−ヒドロキシカルボキサミド(関西大学(Kansai University))、PXD−101(プロリフィックス(Prolifix))、スルホンアミドヒドロキサム酸(メチルジーン(MethylGene))、TPX−HA類似体(CHAP)(ジャパンエナジー(Japan Energy))、オキサムフラチン(Oxamflatin)、トラポキシン(Trapoxin)、デプデシン(Depudecin)、アピジシン(Apidicin)(キョンジ(Kyongji))、ベンズアミド、例えばMS−27−275(三井(Mitsui))、ピロキサミド(Pyroxamide)およびその誘導体、短鎖脂肪酸、例えば酪酸、およびその誘導体、例えば、ピバネックス(Pivanex)(ピバロイルオキシメチルブチレート)、環状テトラペプチド、例えばトラポキシンA、デプシペプチド(FK−228、藤沢(Fujisawa)/NCI)および関連ペプチド化合物、タセジナリン(Tacedinaline)(ファイザー(Pfizer))、MG2856(メチルジーン(MethylGene))、およびHDACクラスIII阻害剤またはSIRT阻害剤(ケリー(Kelly)、オコナー(O’Connor)とマークス(Marks)、2002年、Expert Opin.Inverstig.Drugs 11(12)、p.1695−1713を参照)などの化合物を含むがこれらに限定されないVPAとは異なるヒストンデアチラーゼの阻害剤が挙げられる。
【0053】
VPAまたはその誘導体の投与は、化学療法剤または細胞毒性剤(例えば、5−FU)、分化誘導剤(例えば、ビタミンD、ビタミンD誘導体、レチノイド、イミキモド(imiquimode)などの受容体結合剤)、放射線治療(例えば、エックス線またはガンマ線)、免疫学的方法(抗体治療、ワクチン接種)、併用免疫治療/細胞毒性方法(例えば、細胞毒性成分と結合した抗体)などの投与/適用と組合せることができる。
【0054】
別の実施態様においては、VPAまたはその誘導体は、VPA単独の局所適用によって皮膚腫瘍、基底細胞癌、扁平上皮細胞癌、角化棘細胞腫、ボーエン病、皮膚T細胞リンパ腫、光線性角化症、乾癬、魚鱗癬、座瘡、他の炎症性皮膚疾患を含む腫瘍性および非腫瘍性皮膚疾患における経口適用レチノイド活性を増強する。
【0055】
本発明は、皮膚/粘膜の癌、前悪性皮膚病変、および炎症の局所治療用に単独またはレチノイド、核内受容体リガンド、または5−FUなどの化学療法剤と組合せて使用されるバルプロ酸またはその誘導体を含有する医薬品の局所適用製剤を提供する。
【0056】
それゆえ、本発明の1つの態様は、皮膚/粘膜のさまざまなヒト癌、前悪性皮膚病変、または炎症の局所治療用、および日焼けの治療用に単独もしくはレチノイド、または5−フルオロウラシル(5−FU)と組合せて使用されるVPAまたはその誘導体を含有する局所適用製剤の使用である。したがって、各成分の単独の使用と比べた組合せ局所治療の抗腫瘍活性は増大しうるとともに―必要に応じて―かかる組合せ治療の個別成分の用量は、個別の薬剤単独による治療と関係した望ましくない副作用を減少させるために低下されうる。本発明はまた、上記の疾患の局所治療用の医薬品を製造するためのVPAまたはその誘導体の使用に関する。
【0057】
VPAの誘導体は、式I
【化2】
[式中、R1およびR2は独立して、場合により1個もしくは何個かのヘテロ原子を含んで成り、かつ置換されうる直鎖または分岐、飽和または不飽和、脂肪族C3-25炭化水素鎖であり、R3はヒドロキシル、ハロゲン、アルコキシ、または場合によりアルキル化アミノ基である]によって記載される炭素分岐カルボン酸またはカルボン酸誘導体として定義される。
【0058】
異なるR1およびR2残基はキラル化合物を生じさせる。通常、立体異性体の1つが他より強い催奇形効果を有し(ナウ(Nau)ら、1991年、Pharmacol.Toxicol.69、p.310−321)、催奇形異性体であるほど、より有効にPPARδを活性化する(ランペン(Lampen)ら、1999年)。したがって、この異性体はHDACをより強く阻害することが予想されうる(PCT/EP01/07704号)。本発明は、それぞれの化合物のラセミ混合物、具体的にはより活性の異性体を包含する。
【0059】
炭化水素鎖R1およびR2は、炭化水素鎖で炭素原子を置換する1個もしくは何個かのヘテロ原子(例えば、O、N、S)を含んで成りうる。これは、炭素基のものときわめて類似した構造物が、ヘテロ原子が対応する炭素基と同じ型のハイブリッド形成を有する場合にヘテロ原子基によって採用されうる事実による。
【0060】
R1およびR2は置換されうる。可能な置換基としては、ヒドロキシル基、アミノ基、カルボキシル基、およびアルコキシ基のほか、アリール基および複素環基が挙げられる。
【0061】
好ましくは、R1およびR2は独立して3〜10個、より好ましくは、4〜10個または5〜10個の炭素原子を含んでなる。R1およびR2は独立して飽和されており、または1つの二重結合または1つの三重結合を含んでなる。具体的には、側鎖(R1)の1つは、好ましくは、2および3位でsp1混成炭素原子、または同様の構造を生成するヘテロ原子を含有する。この側鎖は3個の炭素またはヘテロ原子を含んでなるが、長鎖もHDAC阻害分子を生成しうる。また、R2における芳香環またはヘテロ原子の包含は、HDACタンパク質の触媒部位が明らかに多種多様の結合分子を供給するためHDAC阻害活性を有する化合物を生成すると考えられる。催奇性VPA誘導体がHDAC阻害剤であるという所見により、以前に適切な抗てんかん薬とみなされていない化合物もHDAC阻害剤とみなされている(PCT/EP01/07704号)。具体的には、しかし排他的ではなく、R1としてプロピニル残基およびR2として7個もしくはそれ以上の炭素の残基を有する化合物が考えられている(ランペン(Lampen)ら、1999年)。
【0062】
好ましくは、基「COR3」はカルボキシル基である。カルボキシル基の誘導体化も潜在的なHDAC阻害活性を有する化合物の生成が考慮されなければならい。かかる誘導体は、ハロゲン化物(例えば、塩化物)、エステル、またはアミドでありうる。R3がアルコキシである場合、アルコキシ基は1〜25個、好ましくは、1〜10個の炭素原子を含んでなる。R3がモノ−またはジ−アルキル化アミノ基である場合、アルキル置換基は1〜25個、好ましくは、1〜10個の炭素原子を含んでなる。
【0063】
本発明によれば、式Iの化合物の医薬的に許容される塩も製剤用に使用されうる。本発明によれば、人体において式Iで定義される化合物に代謝され、または例えばエステル加水分解によって式Iで定義される化合物の放出をもたらす物質も使用されうる。
【0064】
特定の実施態様では、本発明は、ヒストンデアセチラーゼ活性を有する酵素の阻害剤として式Iに記載されたα−炭素分岐カルボン酸、またはその医薬的に許容される
塩の使用、および癌の局所治療におけるその使用に関し、式中R1は直鎖または分岐、飽和または不飽和、脂肪族C5-25炭化水素鎖であり、R2は独立して直鎖または分岐、飽和または不飽和、脂肪族C2-25炭化水素鎖、ただし、−CH2-CH=CH2、−CH2−C≡CHまたは−CH2−CH2−CH3ではなく、R1およびR2は場合により、ヒドロキシル基、アミノ基、カルボキシル基、アルコキシ基、アリール基、および/または複素環素で置換され、かつR3はヒドロキシルである。
【0065】
さらに別の実施態様では、本発明は、癌の局所治療用の式Iに記載されたα−炭素分岐カルボン酸、またはその医薬的に許容される塩に関し、式中R1は直鎖または分岐、飽和または不飽和、脂肪族C3-25炭化水素鎖であり、R2は独立して直鎖または分岐、飽和または不飽和、脂肪族C3-25炭化水素鎖であり、R1またはR2は炭化水素鎖で炭素原子を置換する1個もしくは何個かのヘテロ原子(例えば、O、N、S)を含んで成り、R1およびR2は場合により、ヒドロキシル基、アミノ基、カルボキシル基、アルコキシ基、アリール基、および/または複素環素で置換され、かつR3はヒドロキシルである。
【0066】
本発明のさらに別の実施態様では、R1およびR2はエステル基(−CO−O−)を含まない。R1およびR2は、ヘテロ原子O、N、またはSを含まない炭化水素鎖でありうる。
【0067】
本発明により使用される最も好ましい化合物は、VPAおよび/または4−ynVPAである。
【0068】
一般に、本発明は、さまざまなヒト疾患を治療する新しい可能性を提供する。出願人は、式Iの化合物のHDAC阻害および細胞分化誘導活性が単独で、または皮膚腫瘍および皮膚T細胞リンパ腫由来の細胞を含む腫瘍細胞の局所治療用のほか、前悪性皮膚病変用、および皮膚/粘膜の炎症性疾患用に既知でかつ臨床的に使用される治療薬と組合せて有効に使用されうることを見出した。組合せ治療は、それらだけで使用される対応する治療薬よりも患者において優れた治療効果を生むと考えられている。本発明の1つの目的は、皮膚に出現する癌の局所治療用に提示された化合物を使用する組合せ治療を提供することである。かかる組合せ治療により、例えば、必要される化学療法薬の治療量の減少がもたらされ、したがって、現在確認される、頻繁に使用される治療薬の部分的にきわめて重篤な副作用を制限しうる。
【0069】
本発明の態様としては、これに限定されないが、現在臨床的使用または臨床的開発における治療原理、例えば、
− 化学療法薬または細胞毒性薬(例えば、5−FU)
− 分化誘発薬剤(例えば、ビタミンD、ビタミンD誘導体、レチノイド、イミキモド(imiquimode)などの受容体結合剤)
− 放射線療法(例えば、エックス線またはガンマ線)
− 免疫学的方法(抗体療法、ワクチン接種)
− 併用免疫治療/細胞毒性法(例えば、細胞毒性成分を結合した抗体)
− 抗血管形成法
と、式Iの化合物との組み合わせを含む。
【0070】
製剤
本発明の組成物は、本発明の組成物が組込まれ、活性剤、および他の任意の活性物質が適切な濃度で皮膚に送達されることを可能にする皮膚科学的に許容される担体を約1%〜約99.9%を含んでなる。
【0071】
担体は、1つもしくはそれ以上の皮膚科学的に許容される固体、半固体、または液体、希釈剤、溶媒、増量剤などを含有しうる。担体は固体、半固体、または液体であってよい。好ましい担体は実質的に半固体である。担体はそれ自体、不活性であり、またはそれ自身の皮膚科学的利点を有しうる。担体の濃度は、選択された担体、活性剤および任意の成分の使用濃度によって変化しうる。
【0072】
適切な担体としては、皮膚科学的に許容される従来の、または他の既知の担体が挙げられる。担体はまた、本明細書に記載された必須成分と物理的および化学的に互換性があり、本発明の組成物と関係がある安定性、有効性、または他の使用利点を過度に損なうものであってはならない。本発明の組成物の好ましい成分は、通常の使用条件下に組成物の有効性を実質的に削減する相互作用がないような方法で混合されることが可能でなければならない。
【0073】
本発明で使用される担体の型は、組成物に所望の製品形態の型によって決まる。本発明において有用な局所組成物は、技術上周知であるような多種多様の製品形態に製造されうる。これらには、ローション、クリーム、ゲル、スティック、スプレー、軟膏、油、泡、粉末、およびペーストが含まれるが、これらに限定されない。これらの製品形態は、溶液、エアロゾル、乳剤、ゲル、固体、およびリポソームを含むが、これらに限定されないいくつかの型の担体を含んで成る。
【0074】
好ましい担体は、皮膚科学的に許容される、親水性希釈剤を含有する。本明細書で使用される「希釈剤」は、特定の材料が分散、溶解、または組込まれうる材料を含む。親水性希釈剤の限定されない例は、水、低級一価アルコール(例えば、C1−C4)、およびプロピレングリコール、ポリエチレングリコール(例えば、分子量200−600g/モル)、ポリプロピレングリコール(例えば、分子量425−2025g/モル)、グリセロール、ブチレングリコール、1,2,4−ブタントリオール、ソルビトールエステル、1,2,6−ヘキサントリオール、エタノール、イソプロパノール、ソルビトールエステル、ブタンジオール、エーテルプロパノール、エトキシル化エーテル、プロポキシル化エーテル、およびその組合せを含む低分子量グリコールおよびポリオールなど有機親水性希釈剤である。水が好ましい希釈である。組成物は、好ましくは、親水性希釈剤を約60%〜約99.99%含んでなる。
【0075】
本発明の溶液は、通常、皮膚科学的に許容される親水性希釈剤を含む。本発明において有用な溶液は、好ましくは、約60%〜約99.99%の親水性希釈剤を含む。
【0076】
本発明によるエアロゾルは、推進剤を上記の溶液に添加することによって形成されうる。例示的な推進剤にはクロロ−フッ化低分子量炭化水素が含まれる。エアロゾルは通常、スプレー式の製品として皮膚に適用される。
【0077】
好ましい担体は、親水性成分、例えば、水または他の親水性希釈剤を含んでなる親水性相、および疎水性成分、例えば、脂質、油、または油性物質を含んでなる疎水性相を含んでなる乳剤を含む。当業者には周知であるように、親水性相は疎水性相中に分散され、または逆もまた同様で、組成物の成分に応じてそれぞれ親水性または疎水性分散および連続相を形成する。乳剤技術においては、「分散相」という語は当業者には周知であり、この相が連続相に懸濁され、または連続相によって囲まれている小さな粒子または液滴として存在することを意味する。分散相は内相または不連続相としても知られている。乳剤は水中油型乳剤、またはシリコーン中水型乳剤など油中水型乳剤であり、または(例えば、三重または他の多相乳剤中に)これを含んで成りうる。水中油型乳剤は通常、約1%〜約50%の分散疎水性相を、かつ約1%〜約98%の連続親水性相を含んで成り、油中水型乳剤は通常、約1%〜約98%の分散親水性相を、約1%〜約50%の連続疎水性相を含んでなる。乳剤は、ゲルネットワークをも含んで成りうる。好ましい乳剤はさらに以下に記載されている。
【0078】
ローションおよびクリームを含むがこれらに限定されない本発明の局所組成物は、皮膚科学的に許容される皮膚軟化剤を含んで成りうる。かかる組成物は、好ましくは、皮膚軟化剤の約2%〜約50%を含有する。皮膚軟化剤は、皮膚をなめらかにする傾向があり、皮膚の平滑性および柔軟性を増大させ、皮膚の乾燥を防止または軽減し、かつ/または皮膚を保護する。皮膚軟化剤は通常、不水溶性、油性、またはロウ状材料である。皮膚軟化剤の非限定的な例が本明細書に記載されている。
【0079】
本発明によるローションおよびクリームは、好ましくは、溶液担体系および1つもしくはそれ以上の皮膚軟化剤を含んでなる。ローションは通常、約1%〜約20%、好ましくは、約5%〜約10%の皮膚軟化剤、約50%〜約90%、好ましくは、約60%〜約80%の水を含んでなる。クリームは通常、約5%〜約50%、好ましくは、約10%〜約20%の皮膚軟化剤、約45%〜約85%、好ましくは、約50%〜約75%の水を含んでなる。
【0080】
本発明の軟膏は、動物性もしくは植物性油または半固体炭化水素(油性)の単純担体基剤、水を吸収して乳剤を形成する吸収軟膏基剤、または水可溶性担体、例えば、水可溶性溶液担体を含んで成りうる。軟膏は、さらに増粘剤を含んで成りうる。例えば、軟膏は約2%〜約10%の皮膚軟化剤、かつ約0.1%〜約2%の増粘剤を含んで成りうる。軟化剤の非限定的な例が本明細書に記載されている。
【0081】
本発明の好ましい局所組成物は乳剤を含んでなる。本発明の乳剤は、以下の1つもしくはそれ以上を含んで成りうる。
【0082】
a)疎水性成分
本発明による乳剤は、脂質、油、油性、または他の疎水性成分を含んでなる疎水性相を含有する。本発明の組成物は、好ましくは、約1重量%〜約50重量%、好ましくは、約1重量%〜約30重量%、かつより好ましくは、約1重量%〜約10重量%の疎水性成分の組成物を含んでなる。疎水性成分は、動物、植物、または石油由来でありうるとともに、天然または合成(すなわち、人工)でありうる。好ましい疎水性成分は実質的に水不溶性、より好ましくは、基本的に水不溶性である。
【0083】
適切な疎水性成分の非限定的な例としては、以下より成る郡から選択されるものが挙げられる。
【0084】
(1)ワセリン液としても知られる鉱油は、石油から得られる液体炭化水素の混合物である。メルクインデックス(The Merck Index)、第10版、登録7048、p.1033(1983年)を参照。
【0085】
(2)ワセリンゼリーとしても知られる鉱油は、非直鎖固体炭化水素および高沸点液体炭化水素のコロイド系であり、液体炭化水素の大部分がミセル内に保持されている。メルクインデックス(The Merck Index)、第10版、登録7047、p.1033(1983年)、シンドラー(Schindler)、Drug.Cosmet.Ind.、89、p.36−37、p.76、p.78−80、p.82(1961年)を参照。
【0086】
(3)約7〜約40個の炭素原子を有する直鎖および分岐鎖炭化水素。これらの炭化水素材料の非限定的な例としては、ドデカン、イソドデカン、スクアラン、コレステロール、硬化ポリイソブチレン、ドコサン、ヘキサデカン、イソヘキサデカンが挙げられる。C7−C40分岐炭化水素、例えば、C13−C14イソパラフィンであるC7−C40イソパラフィンも有用である。
【0087】
(4)直鎖および分岐鎖材料ならびに芳香族誘導体を含むC1−C30カルボン酸およびC2−C30ジカルボン酸のC1−C30アルコールエステル(本明細書では疎水性成分、モノ−およびポリ−カルボン酸に関連して、直鎖、分岐鎖、およびアリールカルボン酸を含む)。非限定的な例としては、セバシン酸ジイソプロピル、アジピン酸ジイソプロピル、ミリスチン酸イソプロピル、パルミチン酸イソプロピル、パルミチン酸メチル、プロピオン酸ミリスチル、パルミチン酸2−エチルヘキシル、ネオペンタン酸イソデシル、マレイン酸ジ−2−エチルヘキシル、パルミチン酸セチル、ミリスチン酸ミリスチル、ステアリン酸ステアリル、ステアリン酸イソプロピル、ステアリン酸メチル、ステアリン酸セチル、ベヘン酸ベヘニル(behenyl behenrate)、マレイン酸ジオクチル、セバシン酸ジオクチル、アジピン酸ジイソプロピル、オクタン酸セチル、ジリノール酸ジイソプロピルが挙げられる。
【0088】
(5)C1−C30カルボン酸のモノ−、ジ−、およびトリ−グリセリド、例えば、カプリル/トリカプリン酸グリセリル、PEG−6カプリル/トリカプリン酸グリセリル、 PEG−8カプリル/トリカプリン酸グリセリル。
【0089】
(6)C1−C30カルボン酸のアルキレングリコールエステル、例えば、エチレングリコールモノ−およびジ−エステル、およびC1−C30カルボン酸のプロピレングリコールモノ−およびジ−エステル、例えば、ジステアリン酸エチレングルコール。
【0090】
(7)前記材料のプロポキシル化およびエトキシル化誘導体。
【0091】
(8)糖および関連材料のC1−C30モノ−およびポリ−エステル。これらのエステルは糖またはポリオール部分および1つもしくはそれ以上のカルボン酸部分由来である。構成酸および糖に応じて、これらのエステルは室温下に液体または固体形態のいずれかである。液体エステルの例としては、テトラオレイン酸グルコース、大豆油脂肪酸のグルコーステトラエステル(不飽和)、混合大豆油脂肪酸のマンノーステトラエステル、オレイン酸のガラクトーステトラエステル、リノール酸のアラビノーステトラエステル、テトラリノール酸キシロース、ペンタオレイン酸ガラクトース、テトラオレイン酸ソルビトール、不飽和大豆油脂肪酸のソルビトールヘキサエステル、ペンタオレイン酸キシリトール、テトラオレイン酸ショ糖、ペンタオレイン酸ショ糖、ヘキサオレイン酸ショ糖、ヘパトオレイン酸ショ糖、オクタオレイン酸ショ糖、およびその混合物が挙げられる。固体エステルの例としては、カルボン酸エステル部分が1:2のモル比でパルミトレイン酸とアラキジン酸であるソルビトールヘキサエステル、カルボン酸エステル部分が1:3のモル比でリノール酸とベヘン酸であるラフィノースのオクタエステル、エステル化カルボン酸部分が、3:4のモル比でヒマワリ種子油脂肪酸とリグノセリン酸であるマルトースのヘプタエステル、エステル化カルボン酸部分が、2:6のモル比でオレイン酸とベヘン酸であるショ糖のオクタエステル、およびエステル化カルボン酸部分が、1:3:4のモル比でラウリン酸とリノール酸とベヘン酸であるショ糖のオクタエステルが挙げられる。好ましい固体材料が、エステル化の程度が7−8であり、不飽和:ベヘル酸のモル比が1:7〜3:5である脂肪酸部分がC18モノ−および/またはジ−不飽和およびベヘン酸であるショ糖ポリエステルである。特に好ましい固体糖ポリステルが、約7個のベヘン脂肪酸部分と約1個のオレイン酸部分が分子中にあるショ糖のオクタエステルである。他の材料としては、ショ糖の綿実油または大豆油脂肪酸エステルが挙げられる。
【0092】
(9)有機ポリシロキサン油。有機ポリシロキサン油は、揮発性、不揮発性、または揮発性と不揮発性のシリコーンの混合物でありうる。この文脈において使用される「不揮発性」という語は、周囲条件下に液体であるシリコーンを指す。この文脈において使用される「揮発性」という語は、他のすべてのシリコーン油を指す。適切な有機ポリシロキサンが、広範囲の揮発度および粘度にわたる多種多様のシリコーンから選択されうる。不揮発性ポリシロキサンが好ましい。適切な有機ポリシロキサン油の例としては、ポリアルキルシロキサン、環状ポリアルキルシロキサン、およびポリアルキルアリールシロキサンが挙げられる。
【0093】
ここでの使用に好ましいのは、ポリアルキルシロキサン、アルキル置換ジメチコーン、シクロメチコーン、トリメチルシルオキシシリケート、ジメチコノール、ポリアルキルアリールシロキサン、およびその混合物から成る群より選択される有機ポリシロキサンである。ここでの使用により好ましいのは、ポリアルキルシロキサンおよびシクロメチコーンである。ポリアルキルシロキサンの中でも好ましいのは、米国特許第5,968,528号明細書に記載されているジメチコーンでである。
【0094】
(10)植物性油および硬化植物性油。植物性油および硬化植物性油の例としては、ヒワマリ油、ヒマシ油、ココナッツ油、綿実油、ニシン油、パーム核油、ヤシ油、ピーナッツ油、大豆油、菜種油、アマニ油、ぬか油、松根油、ゴマ油、ヒマワリ種子油、硬化ベニバナ油、硬化ヒマシ油、硬化ココナッツ油、硬化綿実油、硬化ニシン油、硬化パーム核油、硬化ヤシ油、硬化ピーナッツ油、硬化大豆油、硬化菜種油、硬化アマニ油、硬化ぬか油、硬化ゴマ油、硬化ヒマワリ種子油、およびその混合物が挙げられる。
【0095】
(11)動物油脂、例えば、ラノリンまたはその誘導体、タラ肝油。
【0096】
(12)ポリプロピレングリコールのC4−C20アルキルエーテル、ポリプロピレングリコールのC1−C20カルボン酸エステル、およびジ−C8−C30アルキルエーテルもまた有用である。これらの材料の非限定的な例としては、PPG−14ブチルエーテル、PPG−15ステアリルエーテル、ジオクチルエーテル、ドデシルオクチルエーテルおよびその混合物が挙げられる。
【0097】
b)親水性成分
本発明の乳剤は、親水性成分、例えば、水または他の親水性希釈剤をも含んでなる。したがって、親水性相は、水、または水と1つもしくはそれ以上の水可溶性または分散可能な成分の組合せを含んで成りうる。水を含んでなる親水性成分が好ましい。
【0098】
(c)他の成分
本発明の乳剤および他の局所組成物は、本明細書に記載されているものなどさまざまな他の成分を含んで成る。当業者によって理解されるように、組成物中の成分の親水性に応じて、所定の成分が主に親水性相または疎水性相のいずれかに分散する。
【0099】
本発明の乳剤としては、好ましくは、乳化剤、界面活性剤、構造化剤、および増粘剤から選択される1つもしくはそれ以上の化合物が挙げられる。
【0100】
(1)乳化剤/界面活性剤
乳剤は、連続相内の不連続相を分散および懸濁するのに一般に役立つ乳化剤および/または界面活性剤を含有しうる。多種多様のかかる薬剤が使用されうる。選択された薬剤が化学的および物理的に組成物の基本的な成分と互換性があり、かつ所望の分散特性を示すという条件で、既知または従来の乳化剤/界面活性剤を組成物において使用することができる。適切な薬剤としては、非シリコーン含有乳化剤/界面活性剤、シリコーン乳化剤/界面活性剤、およびその混合物が挙げられる。
【0101】
好ましい実施態様においては、組成物は、親水性乳化剤または界面活性剤を含んでなる。本発明の組成物は、好ましくは、約0.05%〜約5%、より好ましくは、約0.05%〜約1%の少なくとも1つの親水性界面活性剤を含んでなる。
【0102】
好ましい親水性界面活性剤は非イオン界面活性剤から選択される。ここで有用である非イオン界面活性剤は中でも長鎖アルコールの縮合生成物として広範囲に定義されているもの、例えば、糖またはでんぷんポリマーを有する、すなわちグリコシドであるC8−30アルコールである。アルキル基がそれに由来しうる長鎖アルコールの例としては、デシルアルコール、セチルアルコール、ステアリルアルコール、ラウリルアルコール、ミリスチルアルコール、オレイルアルコールなどが挙げられる。これらの界面活性剤の市販の例としては、デシルポリグルコシド(APG 325 CSとしてヘンケル(Henkel)から入手可能)およびラウリルポリグルコシド(APG 600 CSおよび625 CSとしてヘンケル(Henkel)から入手可能)が挙げられる。
【0103】
他の有用な非イオン界面活性剤としては、脂肪酸とのアルキレンオキシドの縮合生成物(すなわち、脂肪酸のアルキレンオキシドエステル)が挙げられる。他の非イオン界面活性剤は、脂肪アルコール酸とのアルキレンオキシドの縮合生成物(すなわち、脂肪アルコールのアルキレンオキシドエーテル)である。さらに他の非イオン界面活性剤は、脂肪酸および脂肪アルコールとのアルキレンオキシドの縮合生成物である[すなわち、ここでポリアルキレンオキシド部分は、一端で脂肪酸によりエステル化されており、もう一端で脂肪アルコールによりエーテル化(すなわち、エーテル連鎖によって結合)されている]。これらアルキレンオキシド由来非イオン界面活性剤の例としては、セテス−6、セテス−10、セテス−12、セテアレス−6、セテアレス−10、セテアレス−12、ステアレス−6、ステアレス−10、ステアレス−12、PEG−6ステアリン酸、PEG−10ステアリン酸、PEG−100ステアリン酸、PEG−12ステアリン酸、PEG−20ステアリン酸グリセリル、PEG−80グリセリル牛脂脂肪酸、PEG−10ステアリン酸グリセリル、PEG−30グリセリルココエート、PEG−80グリセリルココエート、PEG−200グリセリル牛脂脂肪酸、PEG−8ジラウリン酸、PEG−10ジステアリン酸塩、およびその混合物が挙げられる。
【0104】
さらに他の有用な非イオン界面活性剤としては、ポリヒドロキシ脂肪酸アミド界面活性剤が挙げられる。
【0105】
非イオン界面活性剤の中で好ましいのは、ステアレス−21、セテアレス−20、セテアレス−12、ショ糖ココエート、ステアレス−100、PEG−100ステアリン酸塩、およびその混合物から成る群より選択されるものである。
【0106】
この点で使用に適切な他の非イオン界面活性剤としては、糖エステルおよびポリエステル、アルコキシル化糖エステルおよびポリエステル、C1−C30脂肪アルコールのC1−C30脂肪酸エステル、C1−C30脂肪アルコールのC1−C30脂肪酸エステルのアルコキシル化誘導体、C1−C30脂肪アルコールのアルコキシル化エーテル、C1−C30脂肪酸のポリグリセリルエステル、ポリオールのC1−C30エステル、ポリオールのC1−30エーテル、アルキルリン酸塩、ポリオキシアルキレン脂肪エーテルリン酸塩、脂肪酸アミド、乳酸アシル、およびその混合物が挙げられる。これら非シリコン含有乳化剤の非限定的な例としては、ポリエチレングリコール20ソルビタンモノラウレート(ポリソルベート20)、ポリエチレングリコール5大豆ステロール、ステアレス−20、セテアレス−20、PPG−2メチルグルコースエーテルジステアリン酸塩、セテス−10、ポリソルベート80、リン酸セチル、リン酸セチルカリウム、ジエタノールアミンリン酸セチル、ポリソルベート60、ステアリン酸グリセリル、ポリオキシエチレン20トリオレイン酸ソルビタン(ポリソルベート85)、ソルビタンモノラウレート、ポリオキシエチレン4ラウリルエーテルステアリン酸ナトリウム、イソステアリン酸ポリグリセリル−4、ラウリン酸ヘキシル、PPG−2ジステアリン酸メチルグルコースエーテル、PEG−100ステアリン酸塩、およびその混合物が挙げられる。
【0107】
ここで有用な他の乳化剤は、ソルビタンまたはソルビトール脂肪酸エステルおよびショ糖脂肪酸エステルの混合物に基づく脂肪酸エステル混合物である。好ましい脂肪酸エステル乳化剤は、ソルビタンまたはソルビトールC16−C20脂肪酸エステルのショ糖C10−C16脂肪酸エステル、特にソルビタンステアリン酸およびショ糖ココエートとの混合物である。これは、アルラトン(Arlatone)2121の商品名でICIから市販されている。
【0108】
ここで有用な親水性界面活性剤は、代わりとして、または追加として、技術上既知である多種多様の陽イオン、陰イオン、両性イオン、および両性界面活性剤のいずれかを含む。例えば、MaCutcheon’s Detergents and Emulsifiers、North Americann版(1986年)、アルレード出版社(Allured Publishing Corporation)刊を参照。
【0109】
ここで有用な例示的な陽イオン界面活性剤としては、米国特許第5,968,528号明細書に開示されているものが挙げられる。ここで有用な陽イオン界面活性剤としては、四級アンモニウム塩、およびアミノ−アミドなど陽イオンアンモニウム塩が挙げられる。
【0110】
多種多様の陰イオン界面活性剤もここでは有用である。例えば、米国特許第5,968,528号明細書を参照。陰イオン界面活性剤の非限定的な例としては、イセチオン酸アルコイル(例えば、C12−C30)、硫酸アルキルおよびアルキルエーテル、およびその塩、リン酸アルキルおよびアルキルエーテル、およびその塩、タウリン酸アルキルメチル(例えば、C12−C30)、および脂肪酸のせっけん(例えば、アルカリ金属塩、例えば、ナトリウムまたはカリウム塩)が挙げられる。
【0111】
両性イオンおよび両性界面活性剤もここで有用である。本発明の組成物において使用されうる両性イオンおよび両性界面活性剤の例は、脂肪族ラジカルが直鎖または分岐鎖でありうるとともに、脂肪族置換基の1つが約8〜約22個の炭素原子(好ましくは、C8−C18)を含有し、1つが陰イオン水可溶化基、例えば、カルボキシ、スルホン酸塩、硫酸塩、リン酸塩、またはホスホン酸塩を含有する、脂肪族第二級および第三級アミンの誘導体として広範囲に記載されているものである。例えば、アルキルイミノ酢酸塩、およびイミノジアルカン酸塩およびアミノアルカン酸塩、イミダゾリニウム、およびアンモニウム誘導体である。他の適切な両性イオンおよび両性界面活性剤は、ベタイン、スルタイン、ヒドロキシスルタイン、アルキルサルコシン酸塩(例えば、C12−C30)、およびアルカノイルサルコシン酸塩よりなる群から選択されるものである。
【0112】
本発明の好ましい乳剤としては、乳化剤または界面活性剤を含有するシリコーンが挙げられる。多種多様のシリコーン乳化剤がここで有用である。これらのシリコーン乳化剤は通常、当業者にはシリコーン界面活性剤としても知られる有機修飾された有機ポリシロキサンである。有用なシリコーン乳化剤としては、ジメチコーンコポリオールが挙げられる。これらの材料は、ポリエチレンオキシド鎖、ポリプロピレンオキシド鎖、これらの鎖の混合物、およびエチレンオキシドおよびプロピレンオキシド由来の部分を含有するポリエーテル鎖などポリエーテル側鎖を含むように修飾されているポリジメチルシロキサンである。他の例としては、アルキル修飾ジメチコーンコポリオール、すなわち、C2−C30ペンダント側鎖を含有する化合物が挙げられる。さらに他の有用なジメチコーンコポリオールとしては、さまざまな陽イオン、陰イオン、両性イオン、および両性ペンダント部分を有する材料が挙げられる。
【0113】
ここで乳化剤として有用なジメチコーンコポリオールおよび他のシリコーン界面活性剤の非限定的な例としては、ペンダントポリエチレンオキシド側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダントポリプロピレンオキシド側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダント混合ポリエチレンオキシドおよびポリプロピレンオキシド側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダント混合ポリ(エチレン)(プロピレン)オキシド側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダントオルガノベタイン側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダントカルボン酸側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、ペンダント四級アンモニウム側鎖を有するポリジメチルシロキサンポリエーテルコポリマー、かつペンダントC2−C30直鎖、分岐、または環状アルキル部分を含有する前記コポリマーの別の修飾も挙げられる。ダウコーニング社(Dow Corning Corporation)によって販売されているここで有用な市販のジメチコーンコポリオールの例は、ダウコーニング(Dow Corning)(登録商標)190、193、Q2−5220、2501ロウ(Wax)、2−5324流体、および3225Cである(後者の材料は、シクロメチコーンとの混合物として販売されている)。セチルジメチコーンコポリオールは、ポリグリセリル−4イソステアリン酸(および)ラウリン酸ヘキシルとの混合物として市販されており、ABIL(登録商標)WE−09の商品名で販売されている(ゴールドシュミット(Goldschmidt)から入手可能)。セチルジメチコーンコポリオールは、ラウリン酸ヘキシル (および)ポリグリセリル−3オレイン酸(および)セチルジメチコーンとの混合物としても市販されており、ABIL(登録商標)WS−08の商品名で販売されている(やはりゴールドシュミット(Goldschmidt)から入手可能)。ジメチコーンコポリオールの他の非限定的な例としては、ラウリルジメチコーンコポリオール、ジメチコーンコポリオール酢酸塩、ジメチコーンコポリオールアジピン酸塩、ジメチコーンコポリオールアミン、ジメチコーンコポリオールベヘン酸塩、ジメチコーンコポリオールブチルエーテル、ジメチコーンコポリオールヒドロキシステアリン酸塩、ジメチコーンコポリオールイソステアリン酸塩、ジメチコーンコポリオールラウリン酸塩、ジメチコーンコポリオールメチルエーテル、ジメチコーンコポリオールリン酸塩、およびジメチコーンコポリオールステアリン酸塩も挙げられる。
【0114】
ここで有用なジメチコーンコポリオール乳化剤が、例えば、米国特許第5,968,528号明細書に記載されている。
【0115】
(2)構造化剤
この組成物、および特にこの乳剤は、構造化剤を含有しうる。構造化剤は特に本発明の水中油型乳剤において好ましい。理論によって限定されることなく、構造化剤は、組成物の安定性に寄与するレオロジー特性を組成物に提供するのに役立つ。例えば、構造化剤は、液晶ゲルネットワークの形成を補助する傾向がある。構造化剤は、乳化剤または界面活性剤としても機能しうる。本発明の好ましい組成物は、約1%〜約20%、より好ましくは、約1%〜約10%、最も好ましくは、約2%〜約9%の1つもしくはそれ以上の構造化剤を含んでなる。
【0116】
好ましい構造化剤は、約1〜約8個のHLBを有し、かつ少なくとも約45℃の融点を有するものである。適切な構造化剤は、飽和C14〜C30脂肪アルコール、約1〜約5モルのエチレンオキシドを含有する飽和C16〜C30脂肪アルコール、飽和C16〜C30ジオール、飽和C16〜30モノグリセロールエーテル、飽和C16〜C30ヒドロキシ脂肪酸、C14〜C30ヒドロキシル化および非ヒドロキシル化飽和脂肪酸、C14〜C30飽和エトキシル化脂肪酸、アミン、および約1〜約5モルのエチレンオキシドジオールを含有するアルコール、少なくとも40%のモノグリセリド含量を有するC14〜C30飽和グリセリルモノエステル、約1〜約3個のアルキル基および約2〜約3個の飽和グリセロール単位を有するC14〜C30飽和ポリグリセロールエステル、C14〜C30グリセリルモノエーテル、C14〜C30ソルビタンモノ/ジエステル、約1〜約5モルのエチレンオキシドを有するC14〜C30飽和エトキシル化ソルビタンモノ/ジエステル、C14〜C30飽和メチルグルコシドエステル、C14〜C30飽和ショ糖モノ/ジエステル、約1〜約5モルのエチレンオキシドを有するC14〜C30飽和エトキシル化メチルグルコシドエステル、少なくとも約45℃の融点を有する平均1〜2個のグルコース単位を有するC14〜C30飽和ポリグルコシド、およびその混合物から成る群より選択されるものである。
【0117】
本発明の好まし構造化剤は、ステアリン酸、パルミチン酸、ステアリルアルコール、セチルアルコール、ベヘニルアルコール、ステアリン酸、パルミチン酸、平均約1〜約5個のエチレンオキシド単位を有するステアリルアルコールのポリエチレングリコールエーテル、平均約1〜約5個のエンチレンオキシド単位を有するセチルアルコールのポリエチレングリコールエーテル、およびその混合物から成る群より選択される。本発明のより好ましい構造化剤は、ステアリルアルコール、セチルアルコール、ベヘニルアルコール、平均約2個のエチレンオキシド単位(ステアレス−2)を有するステアリルアルコールのポリエチレングリコールエーテル、平均約2個のエチレオキシド単位を有するセチルアルコールのポリエチレングリコールエーテル、およびその混合物から成る群より選択される。さらにより好ましい構造化剤は、ステアリン酸、パルミチン酸、ステアリルアルコール、セチルアルコール、ベヘニルアルコール、ステアレス−2、およびその混合物から成る群より選択される。
【0118】
(3)増粘剤(増粘剤およびゲル化剤を含む)
本発明の組成物はまた、好ましくは、約0.1%〜約5%、より好ましくは、約0.1%〜約3%、および最も好ましくは、約0.25%〜約2%の増粘剤を含んで成る。
【0119】
増粘剤の非限定的なクラスとしては、以下のから成る群より選択される。
【0120】
(i)カルボン酸ポリマー
これらのポリマーは、アクリル酸、置換アクリル酸、およびこれらアクリル酸および置換アクリル酸の塩およびエステル由来の1つもしくはそれ以上のモノマーを含有する架橋化合物であり、ここで架橋剤は2つもしくはそれ以上の炭素−−炭素二重結合を含み、多価アルコール由来である。好ましいカルボン酸ポリマーは、2つの一般的な型である。第1の型のポリマーは、アクリル酸モノマーまたはその誘導体の架橋ホモポリマーである(例えば、ここでアクリル酸は、C1−4アルキル、−−CN、−−COOH、およびその混合物から成る群より独立して選択される2個および3個の炭素位置で置換基を有する)。第2の型のポリマーは、アクリル酸モノマーまたはその誘導体(前文に記載したように)、短鎖アルコール(すなわち、C1−4)アクリレートエステルモノマーまたはその誘導体(例えば、ここでエステルのアクリル酸部分は、C1−4アルキル、−−CN、−−COOH、およびその混合物から成る群より独立して選択される2個および3個の炭素位置で置換基を有する)、およびその混合物から成る群より選択される第1のモノマー、および長鎖アルコール(すなわち、C8−40)アクリレートエステルモノマーまたはその誘導体(例えば、ここでエステルのアクリル酸部分は、C1−4アルキル、−−CN、−−COOH、およびその混合物から成る群より独立して選択される2個および3個の炭素位置で置換基を有する)である第2のモノマーを有する架橋コポリマーである。これら2つの型のポリマーの組合せもここで有用である。
【0121】
第1の型の架橋ホモポリマーにおいて、モノマーは、好ましくは、アクリル酸、メタクリル酸、エタクリル酸、およびその混合物から成る群より選択され、アクリル酸が最も好ましい。第2の型の架橋コポリマーにおいては、アクリル酸モノマーまたはその誘導体は、好ましくは、アクリル酸、メタクリル酸、エタクリル酸、およびその混合物から成る群より選択され、アクリル酸、メタクリル酸、およびその混合物が最も好ましい。短鎖アルコールアクリレートエステルモノマーまたはその誘導体は、好ましくは、C1−4アルコールアクリレートエステル、C1−4アルコールメタクリレートエステル、C1−4アルコールエタクリレートエステル、およびその混合物から成る群より選択され、C1−4アルコールアクリレートエステル、C1−4アルコールメタクリレートエステル、およびその混合物が最も好ましい。長鎖アルコールアクリレートエステルモノマーは、C8−40アルキルアクリレートエステルから選択され、C10−30アルキルアクリレートエステルが好ましい。
【0122】
これらの型のポリマーの両方における架橋剤は、分子当り2個以上のアルケニルエーテル基を含有する多価アルコールのポリアルケニルポリエーテルであり、ここで親多価アルコールは少なくとも3個の炭素原子および少なくとも3個のヒドロキシル基を含有する。好ましい架橋剤は、ショ糖のアリルエーテルおよびペンタエリトリトールのアリルエーテル、およびその混合物から成る群より選択されるものである。
【0123】
ここで有用な第1の型の市販のホモポリマーの例としては、ショ糖またはペンタエリトリトールのアリルエーテルを有するアクリル酸架橋のホモポリマーであるカルボマーである。カルボマーは、カルボポール(Carbopol)(登録商標)900シリーズとしてB.F.グッドリッチ(Goodrich)から入手可能である(例えば、カルボポール(Carbopol)(登録商標)954)。ここで有用な第2の型の市販のコポリマーの例としては、アクリル酸、メタクリル酸、またはその短鎖(すなわち、C1−4アルコール)エステルの1つのモノマーの1つもしくはそれ以上を有するC10−30アルキルアクリレートのコポリマーであり、ここで架橋剤はショ糖またはペンタエリトリトールのアリルエーテルである。これらのコポリマーは、アクリレート/C10−30アルキルアクリレートクロスポリマーとして知られており、カルボポール(Carbopol)(登録商標)1342、カルボポール(Carbopol)(登録商標)1382ペムレン(Pemulen)TR−1、およびペムレン(Pemulen)TR−2として、B.F.グッドリッチ(Goodrich)から市販されている。言い換えれば、ここで有用なカルボン酸ポリマー増粘剤の例は、カルボマー、アクリレート/C10−30アルキルアクリレートクロスポリマー、およびその混合物から成る群より選択されるものである。
【0124】
(ii)架橋ポリアクリレートポリマー
増粘剤またはゲル化剤として有用な架橋ポリアクリレートポリマーとしては、陽イオンおよび非イオンポリマーが挙げられ、陽イオンポリマーが一般に好ましい。適切な架橋ポリアクリレートポリマーの非限定的な例は、米国特許第5,968,528号明細書に開示されている。
【0125】
(iii)ポリアクリルアミドポリマー
ポリアクリルアミドポリマー、特に、置換分岐または非分岐ポリマーを含む非イオンポリアクリルアミドポリマーもここで有用である。これらのポリマーは、1個または2個のアルキル基(好ましくは、C1〜C5)で置換されていない、または置換されているアクリルアミドおよびメタクリルアミドを含むさまざまなモノマーから形成されうる。アミド窒素が置換されていない、または1個または2個のC1〜C5アルキル基(好ましくは、メチル、エチル、またはプロピル)で置換されているアクリレートアミドおよびメタクリレートアミドモノマー、例えば、アクリルアミド、メタクリルアミド、N−メタクリルアミド、N−メチルメタクリルアミド、N,N−ジメチルメタクリルアミド、N−イソプロピルアクリルアミド、N−イソプロピルメタクリルアミド、およびN,N−ジメチルアクリルアミドが好ましい。これらのポリマーは、約1,000,000より大きい、好ましくは、約1,5000,000よりも大きい、かつ約30,000,000までの分子量を有する。これらのポリアクリルアミドポリマーの中で最も好ましいのは、CTFAで表示されるポリアクリルアミドである非イオンポリマーおよびイソパラフィンおよびラウレス−7(laureth−7)であり、これはセピゲル(Sepigel)305の商品名でセピック社(Seppic Corporation)(ニュージャージー州(N.J.)、フェアフィールド(Fairfield))から入手可能である。
【0126】
ここで有用な他のポリアクリルアミドポリマーとしては、アクリル酸および置換アクリル酸を有するアクリルアミドおよび置換アクリルアミドの多ブロックコポリマーが挙げられる。これらの多ブロックコポリマーの市販の例としては、リポケミカルズ社(Lipo Chemicals,Inc.)(ニュージャージー州(N.J.)、パターソン(Patterson))のハイパン(Hypan)SR150H、SS500V、SS500W、SSSA100Hが挙げられる。
【0127】
(iv)多糖類
多種多様の多糖類がここで有用である。「多糖類」によって、繰り返し糖(すなわち、糖質)単位の骨格を含有するゲル化剤が意味される。多糖類ゲル化剤の非限定的な例としては、セルロース、カルボキシメチルヒドロキシエチルセルロース、セルロースアセテートプロピオン酸カルボキシレート、ヒドロキシエチルセルロース、ヒドロキシエチルエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、メチルヒドロキシエチルセルロース、微結晶性セルロース、硫酸ナトリウムセルロース、およびその混合物から成る群より選択されるものである。ここではアルキル置換セルロースも有用である。これらのポリマーにおいては、セルロースポリマーのヒドロキシ基は、ヒドロキシアルキル化(好ましくは、ヒドロキシエチル化またはヒドロキシプロピル化)されてヒドロキシアルキル化セルロースを形成し、これは次いでエーテル連鎖によりC10−C30直鎖または分岐鎖アルキル基で修飾される。通常、これらのポリマーは、ヒドロキシアルキルセルロースを有するC10−C30直鎖または分岐鎖アルコールのエーテルである。ここで有用なアルキル基の例としては、ステアリル、イソステアリル、ラウリル、ミリスチル、セチル、イソセチル、ココイル(すなわち、ココナッツ油のアルコール由来のアルキル基)、パルミチル、オレイル、リノレイル、リノレニル、リシノレイル、ベヘニル、およびその混合物から成る群より選択されるものが挙げられる。中でも好ましいアルキルヒドロキシアルキルセルロースエーテルは、CTFAで表示される材料であるセチルヒドロキシエチルセルロースであり、これはセチルアルコールおよびヒドロキシエチルセルロースのエーテルである。この材料は、ナトロソール(Natrosol)(登録商標)CSプラスの商品名でアクアロン社(Aqualon Corporation)から市販されている。
【0128】
v)ゴム
ここで有用な他の追加の増粘化およびゲル化剤としては、主に天然源由来である材料が挙げられる。これらのゲル化剤ゴムの非限定的な例としては、アカシア、寒天、アルギン、アルギン酸、アルギン酸アンモニウム、アミロペクチン、アルギン酸カルシウム、カラギーナンカルシウム、カルニチン、カラギーナン、デキストリン、ゼラチン、ゲランゴム、グアーガム、グアーヒドロキシプロピルトリモニウムクロリド、ヘクトライト、ヒアルロン酸、ケイ酸、ヒドロキシプロピルキトサン、ヒドロキシプロピルグアー、カラヤゴム、昆布、ローカストビーンゴム、納豆ゴム、アルギン酸カリウム、カリウムカラギーナン、アルギン酸プロピレングリコール、スクレロチウムゴム、カルボイキシメチルデキストランナトリウム、カラギーナンナトリウム、トラガカントゴム、キサンタンガム、およびその混合物から成る群より選択される材料が挙げられる。
【0129】
(vi)架橋ビニルエーテル/無水マレイン酸コポリマー
ここで有用な他の追加の増粘化およびゲル化剤としては、アルキルビニルエーテルおよび無水マレイン酸の架橋コポリマーが挙げられる。
【0130】
(vii)架橋ポリ(N−ビニルピロリドン)
追加の増粘化およびゲル化剤としてここで有用な架橋ポリビニル(N−ピロリドン)。これらのゲル化剤は通常、約2〜約12個の炭素原子を含有する末端ジオールのジビニルエーテルおよびジアリルエーテル、約2〜約600単位を含有するポリエチレングリコールのジビニルエーテルおよびジアリルエーテル、約6〜約20個の炭素原子を有するジエン、ペンタエリトリトールのジビニルベンゼン、ビニル、およびアリルエーテルなどから成る群より選択される架橋剤約0.25〜約1重量%を含有する。
【0131】
本発明の好ましい組成物としては、カルボン酸ポリマー、架橋ポリアクリレートポリマー、ポリアクリルアミドポリマー、およびその混合物から成る群より選択され、より好ましくは、架橋ポリアクリレートポリマー、ポリアクリルアミドポリマー、およびその混合物から成る群より選択される増粘剤が挙げられる。
【0132】
任意の成分
本発明の局所組成物は多種多様の任意の成分を含んで成りうるが、かかる任意の成分は物理的および化学的に本明細書に記載された基本的な成分と互換性があり、本発明の組成物と関係した安定性、有効性、または他の使用利点を過度に損なうことがないことを条件とする。任意の成分を、本組成物の担体中に分散、溶解などすることができる。
【0133】
任意の成分としては、エステ剤および活性剤が挙げられる。例えば、組成物としては、本発明の基本的な成分に加えて、吸収剤(粘土など油吸収剤およびポリマー吸収剤を含む)、研磨剤、アンチケーキング剤、消泡剤、抗菌剤(例えば、微生物を破壊し、微生物の成長を阻止し、または微生物の病原性作用を阻止することが可能であり、例えば、座瘡の制御および/または局所組成物の保存に有用である化合物)、結合剤、生物学的添加剤、緩衝剤、充填剤、化学添加剤、化粧殺生剤、変性剤、化粧収斂剤、薬剤収斂剤、外用鎮痛薬、被膜剤、保湿剤、乳白剤、芳香剤、香料、顔料、着色剤、精油、皮膚知覚剤(skin sensate)、皮膚軟化剤、皮膚緩和剤、皮膚治療薬、pH調整剤、可塑剤、防腐剤、防腐増強剤、推進剤、還元剤、皮膚調整剤、皮膚透過増強剤、皮膚保護剤、溶媒、懸濁剤、乳化剤、増粘剤、可溶化剤、組成物の被膜特性および実質性を補助するためのポリマー(例えば、エイコセンス(eicosense)およびビニルピロリドンのコポリマーであり、その例はGAFケミカル社(Chemical Corp)からガネックス(Ganex)(登録商標)V−220として入手可能)、ロウ、日焼け止め剤(sunscreen)、日焼け止め剤(sunblock)、紫外線吸収剤または散乱剤、サンレスタンニング剤、抗酸化剤および/またはラジカルスカベンジャー、キレート化剤、金属イオン封鎖剤、抗座瘡剤、抗炎症剤、抗男性ホルモン物質、脱毛剤、落屑剤/エクスフォリアント、有機ヒドロキシ酸、ビタミン、およびその誘導体(ビタミンCおよびリン酸アスコルビルなど水分散性または可溶性ビタミンを含む)、コラーゲン産生を刺激する化合物、および天然抽出物を挙げることができる。他のかかる材料は技術上既知である。かかる材料の非排他的な例は、Pharmaceutical Dosage Forms−Disperse Systems、リーバーマン(Lieberman)、リーガー・アンド・バンカー(Riger & Banker)、第1巻(1988年)および第2巻(1989年)、または米国特許第5,968,528号明細書に記載されている。
【0134】
本発明の組成物は、皮膚軟化剤を含んで成りうる。皮膚軟化剤は、以下のクラスの1つもしくはそれ以上から選択されうる。すなわち、植物性および動物性油、およびヒマシ油、カカオバター、ベニバナ油、綿実油、トウモロコシ油、オリーブ油、タラ肝油、アーモンド油、アボガド油、ヤシ油、ゴマ油、スクアレン、キクイ(kikui)油、および大豆油を含むがこれらに限定されないトリグリセリド、アセチル化モノグリセリドなどアセトグリセリドエステル、エトキシル化グリセリルモノステアリン酸などエトキシル化グリセリド、メチル、イソプロピル、およびラウリン酸ヘキシル、ラウリン酸イソヘキシル、パルミチン酸イソヘキシル、パルミチン酸イソプロピル、パルミチン酸メチル、オレイン酸デシル、オレイン酸イソデシル、ステアリン酸ヘキサデシル、ステアリン酸デシル、イソステアリン酸イソプロピル、イソステアリン酸メチル、アジピン酸ジイソプロピル、アジピン酸ジイソヘキシル、アジピン酸ジヘキシルデシル、セバシン酸ジイソプロピル、乳酸ラウリル、乳酸ミリスチル、乳酸セチルなどの脂肪酸のブチルエステルを含むがこれらに限定されない、10〜20個の炭素原子を有する脂肪酸のアルキルエステル、ミリスチン酸オレイル、ステアリン酸オレイル、およびオレイン酸オレイルなど10〜20個の炭素原子を有する脂肪酸のアルケニルエステル、ペラルゴン酸、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、イソステアリン酸、ヒドロキシステアリン酸、オレイン酸、リノール酸、リシノール酸、アラキン酸、ベヘン酸、およびエルカ酸などの10〜20個の炭素原子を有する脂肪酸、ラウリル、ミリスチル、セチル、ヘキサデシル、ステアリル、イソステアリル、ヒドロキシステアリル、オレイル、リシンオレイル、ベヘニル、エルシル、および2−オクチルドデカニルアルコールなどの10〜20個の炭素原子を有する脂肪アルコール、ラノリン、ラノリン油、ラノリンロウ、ラノリンアルコール、ラノリン脂肪酸、ラノリン酸イソプロピル、エトキシル化コレステロール、プロポキシル化ラノリンアルコール、アセチル化ラノリンアルコール、リノレイン酸ラノリンアルコール、リシンオレイン酸ラノリンアルコール、リシンオレイン酸ラノリンアルコールの酢酸塩、エトキシル化アルコール−エステルの酢酸塩、ラノリンの水素添加分解、エオキシル化硬化ラノリン、および液体および半固体ラノリン吸収基剤などラノリンおよびラノリン誘導体、エチレングリコールモノおよびジ−脂肪酸エステル、ジエチレングリコールモノ−およびジ−脂肪酸エステル、ポリエチレングリコール(200−6000)モノ−およびジ−脂肪酸エステル、プロピレングリコールモノ−およびジ−脂肪酸エステル、ポリプロピレングリコール2000モノオレイン酸、ポリプロピレングリコール2000モノステアリン酸、エトキシル化プロピレングリコールモノステアリン酸、グリセリルモノ−およびジ−脂肪酸エステル、ポリグリセロールポリ脂肪エステル、エトキシル化グリセリルモノステアリン酸、1,2−ブチレングリコールモノステアリン酸、1,2−ブチレングリコールジステアリン酸塩、ソルビタン脂肪酸エステル、およびポリオキシエチレンソルビタン脂肪酸エステルなどの多価アルコールエステル、蜜ロウ、鯨ろう、ミリスチン酸ミリスチル、ステアリン酸ステアリルなどのろうエステル、エーテルエステルの混合物を形成する各種エチレンオキシド含量のエトキシル化ソルビトールとの蜜ロウの反応生成物であるポリオキシエチレンソルビトール蜜ロウなどの蜜ロウ誘導体、カルナバおよびカンデリラロウを含むがこれらに限定されない植物ロウ、レシチンおよび誘導体などリン脂質、コレステロールおよびコレステロール脂肪酸エステルを含むがこれらに限定されないステロール、および脂肪酸アミド、エトキシル化脂肪酸アミド、および固体脂肪酸アルカノールアミドなどのアミド。
【0135】
組成物は、例えば、多価アルコール型の保湿剤を含んで成りうる。代表的な多価アルコールとしては、ポリアルキレングリコール、およびより好ましくは、プロピレングリコール、ジプロピレングリコール、ポリプロピレングリコール、ポリエチレングリコールおよびその誘導体、ソルビトール、ヒドロキシプロピルソルビトール、エリトリトール、トレイトール、ペンタエリトリトール、キリシトール、グルシトール、マンニトール、ヘキシレングリコール、ブチレングリコール(例えば、1,3ブチレングリコール)、ヘキサントリオール(例えば、1,2,6−ヘキサントリオール)、グリセロール、エトキシル化グリセロール、プロポキシル化リセロール、2−ピロリドン−5−カルボン酸ナトリウム、可溶性コラーゲン、フタル酸ジブチル、ゼラチンおよびこれらの混合物を含むアルキレンポリオールおよびその誘導体が挙げられる。
【0136】
別の任意の成分は、グアニジン;グリコール酸およびグリコール酸塩(例えば、アンモニウムおよび四級アルキルアンモニウム);乳酸および乳酸塩(例えば、アンモニウムおよび四級アルキルアンモニウム);そのさまざまな形態のいずれかにおけるアロエ(例えば、アロエゲル);糖およびでんぷん誘導体(例えば、アルコキシル化グルコース);ヒアルロン酸およびその誘導体(例えば、ヒアルロン酸ナトリウムなど塩誘導体);ラクトアミドモノエタノールアミン、アセトアミドモノエタノールアミン;尿素;パンテノール;糖;でんぷん;シリコーン液;シリコーンゴム;およびそれらの混合物である。プロポキシル化グリセロールも有用である。
【0137】
親水性および親油性のクリームが調査された。これらのクリームは、高生体適合性成分で構成されている。具体的には、これのクリーム系の脂質相は、オリーブ油、甘扁桃油、アボカド油、ホホバ油およびその他などの任意の植物性油、ワセリンまたはその他など鉱油、および/または脂肪酸のエステル、短、中、長鎖トリグリセリドなどの任意の合成油相で構成されうる。これらの系を調製するために、クリームに特定の物理化学的安定性を提供することが可能である皮膚科学的に許容される界面活性剤、例えば、レシチン、トゥイーン(Tween)80などが使用されうる。ゲル系製剤もバルプロ酸、その塩および誘導体の局所適用に可能性のある製剤として調査された。製剤に所望のレオロジー特性を提供することが可能である増粘剤を使用することによって、ゲルを調製することができる。この場合にも、ゲル系製剤の調製に使用される薬剤は、特定のヒト皮膚寛容性、すなわち、セピゲル(sepigel)、ジルゲル(zilgel)、カルボポール(carbopol)、キサンタンゴム、ゼラチン、PVP、PEOなどを有する必要がある。
【0138】
投与
特定の患者用の特定の局所用量レベルは、年齢、体重、一般的健康状態、性別、食習慣、および投薬歴、および患者の特定の疾患の重篤度、および使用される特定の化合物の活性、投与時間、排泄率、治療期間、他の薬剤、化合物、および/または組合せて使用される材料を含むさまざな因子に応じて用いることができる。活性化合物の適切な投与量、および活性化合物を含んでなる組成物が、患者によって変動することは理解されるであろう。最適な投与量の決定は、一般に本発明の治療のリスクまたは有害な副作用に対する治療的有用性のレベルの平衡を含む。
【0139】
インビボでの局所投与は、治療過程全体を通じて1回量、連続的、または断続的に達成されうる。投与の最も有効な手段および投与量を決定する方法は当業者に周知であり、治療に使用される製剤、治療の目的、治療される標的細胞、および治療される対象によって変動する。1回または多数の投与が、治療医師によって選択される用量レベルおよびパターンで実施されうる。
【0140】
一般に、適切な用量の活性化合物、すなわち、VPAまたはVPA誘導体は、少なくとも約0.1重量%、好ましくは、0.1重量%〜6重量%、より好ましくは、0.3重量%〜5重量%、より好ましくは、0.5重量%〜4重量%、かつさらにより好ましくは、1重量%〜4重量%の濃度を含んで成り、活性成分は医薬的に許容される担体に懸濁される。活性成分が塩、エステル、プロドラッグなどである場合、投与される量は親化合物に基づき計算され、こうして使用される実重量は比例して増大する。
【0141】
活性化合物、すなわち、VPAまたはVPA誘導体がレチノイドと組合せて使用される場合は、本活性化合物の適切な用量は、少なくとも0.1重量%、好ましくは、0.1重量%〜6重量%、より好ましくは、0.3重量%〜5重量%、より好ましくは、0.5重量%〜4重量%、かつさらにより好ましくは、1重量%〜4重量%の範囲であり、使用されるレチノイドについては、活性化合物の適切な用量は約0.01重量%〜約1重量%、好ましくは、0.05重量%〜0.5重量%の範囲である。
【0142】
活性化合物、すなわち、VPAまたはVPA誘導体が5−フルオロウラシルと組合せて使用される場合は、本活性化合物の適切な用量は、少なくとも0.1重量%、好ましくは、0.1重量%〜6重量%、より好ましくは、0.3重量%〜5重量%、より好ましくは、0.5重量%〜4重量%、かつさらにより好ましくは、1重量%〜4重量%の範囲であり、使用される5−フルオロウラシルについては、活性化合物の適切な用量は約0.1重量%〜約10重量%、好ましくは、1重量%〜10重量%の範囲である。
【0143】
本発明の好ましい実施態様においては、VPAは、毎日1回〜3回、蓄積総1日量0.5mg(ミリグラム)〜10mg(ミリグラム)で約1cm2(qcm)の病変当りに局所使用される。より好ましい実施態様においては、VPAは、毎日1回〜3回、蓄積総1日量1mg(ミリグラム)〜8mg(ミリグラム)で約1cm2(qcm)の病変当りに局所使用される。さらにより好ましい実施態様においては、VPAは、毎日1回〜3回、蓄積総1日量2mg(ミリグラム)〜6mg(ミリグラム)で約1cm2(qcm)の病変当りに局所使用される。実際の1日量は、治療される病変のサイズによって決まり、したがって、1日量の増大に従って約100cm2(qcm)までの病変サイズを含めて、病変サイズの増大とともに増加する。
【0144】
本発明の他の好ましい実施態様においては、VPAを含有する局所製剤は、リストアップされた成分(含量は%[w/w]で示されている)を含有する以下の3つの製剤によって構成されている。
【0145】
製剤例1:
白鉱油 20
セチルアルコール 24
セトマクロゴール1000 6
バルプロ酸 >0.1、例えば、0.1〜6
白ワセリン 100に添加
【0146】
製剤例2:
酸化亜鉛 25
でんぷん 25
バルプロ酸 >0.1、例えば、0.1〜6
白ワセリン 100に添加
【0147】
製剤例3:
白ワセリン 25
セチルアルコール 10
トゥイーン(Tween)60 5
グリセリン 10
EDTA 0.2
香水 (場合により) 2滴
バルプロ酸ナトリウム >0.1、例えば、0.1〜6
再蒸留水 100に添加
【実施例】
【0148】
実施例1
VPAまたはレチノイン酸誘導体タザロテン単独、およびVPAとタザロテンの組合せによる治療時のヒト不死化ケラチン生成細胞(HaCaT細胞)における相乗的細胞増殖阻害およびアポトーシスの誘導(図1および5)。
【0149】
方法:
MTTアッセイ:この比色アッセイでは存在する細胞の数に比例するホルマザンの形成を検出する。薬剤を含有する組織培養培地を除去し、2mg/ml MTT((3−4,5−ジメチルチアゾール−2イル)臭化2,5−ジフェニルテトラゾリウムブロミド)を含有するDME 200μlを各ウェルに添加した。96ウェルプレートを37℃、10%CO2下に30分間インキュベートした。次いで、MTTを含有する培養培地を除去し、細胞をPBSで1回洗浄した。PBSの除去後、200μL DMSOを各ウェルに添加し、プレートを回転プラットフォームシェーカー上に5分間配置した。分光光度計を使用して550nmの波長で吸光度を測定することによって結果を得た。結果は、対照を100%とするパーセンテージで示されている。
【0150】
アポトーシスアッセイ:細胞を含有するSub−G1 DNAのパーセンテージを分析することでベクトン・ディッキンソン(Beckton Dickinson)FACScaliburフローサイトメトリーによってアポトーシスを測定した。10000個のイベントが各サンプルで記録された。増幅スケールはすべてのパラメータで線形であった。光電子増倍管電圧を、FL2−H対SCC−Hグラフにおけるチャンネル300でG0/G1細胞に相当するピークDNA含量を配置するように設定した。ピークDNAよりも低い含量を示す細胞(低二倍体(hypodiploid)細胞)および高SSC−H(顆粒高凝縮細胞)をアポトーシスとみなした。実験を2通りで3回繰り返した。エラーバーは1標準偏差を示す。
【0151】
結果:
図1は、表示された化合物で3日間処理したヒト不死化ケラチン生成細胞(HaCaT細胞)で行われた増殖アッセイの結果をグラフで示す。薬剤をプレーティングと同じ日に添加した(0日目)。MTTアッセイ(細胞増殖を測定するために)を3日目に行った。薬剤のさまざまな組合せを増殖アッセイで使用した。図に示すように、単独で使用されたVPAおよびタザロテンは細胞増殖を阻害し、タザロテン+ヒストンデアセチラーゼ阻害剤VPAの組合せは、MTTアッセイによって測定されるように、両方の薬剤の単独の阻害よりも細胞増殖の阻害でより有効であった。
【0152】
同様に、VPAはHaCaT細胞におけるタザロテン誘発アポトーシスを増強する(図5)。細胞を0日目に直径35mmのウェルにプレーティングした。薬剤を1日目に添加した。アポトーシスアッセイを2日目に行った。VPAは、subG1 DNA含量の明らかな増大を誘発し、これがヒト不死化ケラチン生成細胞HaCaTのアポトーシスを誘発することを示した。図5に示されているように、タザロテン+VPAの組合せは、単独で使用された両方の薬剤で確認されたアポトーシス率よりもアポトーシスの誘発でより有効であった。したがって、これら2つの薬剤の組合せ活性は、この処理の相乗的活性によって説明されなければならない。
【0153】
実施例2
VPAまたはレチノイン酸(RA)単独、およびVPAとレチノイン酸(RA)の組合せによる処理でのヒト不死化ケラチン生成細胞(HaCaT細胞)における相乗的細胞増殖阻害およびアポトーシスの誘導(図2)。
【0154】
方法:
MTTアッセイ:詳細については実施例1を参照。
【0155】
結果:
図2に示されているように、単独で使用されたVPAおよびRAはヒトケラチン生成細胞HaCaTの増殖を阻害し、両方の薬剤を組合せて使用すると、VPAはRA誘発細胞増殖阻害をさらに増強した。細胞を0日目にウェルにプレーティングした。次いで、薬剤を指示濃度で添加した(VPA1+RA1:VPA 1mM+レチノイン酸1μM、VPA1+RA2:VPA 1mM+レチノイン酸2μM)。MTTアッセイを3日目に行った。薬剤のさまざまな組合せを増殖アッセイでも使用した。RA+ヒストンデアセチラーゼ阻害剤VPAの組合せは、MTTアッセイによって測定されるように、単独で使用された各薬剤よりも細胞増殖の阻害でより有効でった。したがって、これら2つの薬剤の組合せ活性は、この治療の相乗的活性によって説明されなければならない。
【0156】
実施例3
VPAまたは5−フルオロウラシル単独、およびVPAと5−フルオロウラシル(5−FU)の組合せによる処理でのヒト不死化ケラチン生成細胞(HaCaT細胞)における相乗的細胞増殖阻害およびアポトーシスの誘導(図3)。
【0157】
方法:
MTTアッセイ:詳細については実施例1を参照。
【0158】
結果:
単独で使用されたVPAおよび5−FUはHaCaT細胞の増殖を阻害し、VPAは、両方の薬剤を組合せて使用すると、これらの細胞における5−FU誘発増殖阻害を増強した(図3)。薬剤をプレーティングと同じ日に添加した(0日目)。MTTアッセイ(細胞増殖を測定するために)を3日目に行った。5μMまたは10μM 5−FUおよび1mM VPAを使用した。5−FU+HDAC阻害剤VPAの組合せは、MTTアッセイによって測定されるように、単独で使用された各薬剤よりも細胞増殖の阻害でより有効でった。したがって、これら2つの薬剤の組合せ活性は、この治療の相乗的活性によって説明されなければならない。
【0159】
実施例4
VPAは、ヒト不死化ケラチン生成細胞(HaCaT細胞)における細胞周期停止を誘発する(図4)。
【0160】
方法:
BrdU取込みアッセイ:細胞増殖をBrdUで細胞を標識化することによって検出した。指示濃度でVPAによる処理の24時間後、メーカー(ZYMED、カリフォルニア州(CA)、サンフランシスコ(San Francisco))のプロトコールに従いBrdU(1:100)で細胞をインキュベートした。細胞を1時間、培地中でインキュベートし、次いで70%エタノールで固定した。取込みBrdUを間接免疫ペルオキシダーゼ法(アマシャム(Amersham)、イリノイ州(IL)、アーリントンハイツ(Arlington Heights))。手短に言えば、培養細胞を最初に1時間、ビオチン−マウス抗BrdU抗体でインキュベートした。TTBS緩衝液(20mMトリス(Tris)、500mM NaCl、0.05%トゥイーン(Tween)−20、pH7.5)中で洗浄後、細胞をさらにビオチン化ヤギ抗マウス免疫グロブリンで約10分間インキュベートした。次いで、細胞を洗浄し、酵素(ペルオキダーゼ)複合体で10分間、室温下にインキュベートした。基質−色原体の添加によって免疫反応を示した。陽性細胞を無作為に選んだ10箇所の顕微鏡視野で計算した。
【0161】
結果:
HaCaT細胞を直径35mmのウェルに0日目にプレーティングした。VPAを指示濃度で0日目に添加した。BrdUアッセイを2日目に行った。図4に示されているように、VPAはBrdU陽性細胞の用量依存減少を引き起こし、これが細胞周期停止および/またはヒトケラチン生成細胞の細胞生存の喪失を誘発することを示した。
【0162】
実施例5
3%(w/w)で局所適用されるVPAは、時間依存様態でヒト皮膚に透過する(図6)。
【0163】
方法:
局所適用可能な水性VPA製剤の調製:セピゲル(Sepigel)(セピック(Sepic)、イタリア、ミラノ(Milan))(2%w/w)およびバルプロ酸ナトリウム(3%w/w)を適量の蒸留水中に分散させ、室温下に持続的に攪拌しながら局所バルプロ酸ナトリウム製剤を調製した。結果として生じるゲルを使用前の24時間、室温下に保存した。
【0164】
インビトロ皮膚透過アッセイ:表皮角質層(SCE)を有する細胞膜でアセンブリしたフランツ(Franz)型ガラス拡散セル(LGA、米国、カリフォルニア州(CA)、バークリー(Berkeley))を使用して分析を行った。成人ヒト皮膚サンプル(平均年齢29±5歳)を腹部減量手術から得た。皮下脂肪を切除し、皮膚サンプルを60℃下の蒸留水中に1分間、浸漬し、次いで表皮角質層(SCE)を剥離した。SCEサンプルを約25%相対湿度下に維持した乾燥機で室温下に乾燥させた。乾燥サンプルをアルミニウムホイルに包み、使用まで4℃下に保存した。乾燥SCEのサンプルをフランツ(Franz)型拡散セルに載せられる前の1時間、室温下、蒸留水中に浸漬することによって再水和した。露出皮膚表面積は0.75cm2であり、受容体容積は4.5mlであった。受入溶液、NaCl0.9%(w/w)の溶液を攪拌し、実験中に35℃下に自動温度調節した。このプロトコールで使用されるSCE膜の皮膚バリアの完全性をそのトリチウム水透過係数(Kp)を測定することによって評価した。Kp値は、1.58±0.3×10-3cmh-1であることがわかり、対応する文献において以前に報告されたものと一致した(ブロナウ(Bronaugh)ら、1986年、J Invest Dermatol 87、p.451−453)。バルプロ酸ナトリウム(バルプロエート(Valproate))を3%(w/w)で皮膚表面に局所適用した。各実験を2通りで3種類の皮膚ドナーで行った。受入溶液のサンプルを実験期間中(10時間)の異なる時点で回収し、サンプル量を同量の新しい溶液に取り替えた。全サンプルを分光蛍光検出とともにHPLCによってバルプロ酸ナトリウム含量について分析した。結果を透過総量(mg)で表した。
【0165】
バルプロ酸ナトリウムの定量:バルプロ酸ナトリウムを測定するために、4−ブロモメチル−7−メトキシクマリン(BMMC)でサンプルを誘導体化した。シクロヘキサンカルボン酸を内部標準として使用した。誘導体化は以前に報告されたように行った(ブスケ(Bousquet)ら、1991年、Pharmazie 46、p.257−258)。分析は、20μlレオダイン(Rheodyne)モデル7125注入弁(レオダイン(Rheodyne)、カリフォルニア州、コタチ(Cotati))を取付けた1050ヒューレットパッカード(Hewlett Packard)液体クロマトグラファーから構成されたクロマトグラフィーシステムで行われた。クロマトグラフィー分析は、ハイパーシル(Hypersil)ODS 5μmカラム(ポリコンサルト(Policonsult)、イタリア、ローマ(Rome))で行われた。移動相は、流量1ml/分でCH3CN/H2O(65/35v/v)であった。検出は310nmで行われた。
【0166】
結果:
図6は、インビトロ透過アッセイの結果を示す。成人ヒト皮膚サンプル(皮膚表面積は0.75cm2であった)を方法の部に記載したように調製した。適量のバルプロ酸ナトリウム(バルプロエート(Valproate))を3%(w/w)でフランツ(Franz)型ガラス拡散セルに配置した皮膚に局所適用した。各実験を2通りで3種類の異なる皮膚ドナーで行った。受入溶液のサンプルを実験期間中(10時間)の異なる時点で回収し、サンプル量を同量の新しい溶液に取り替えた。全サンプルを分光蛍光検出とともにHPLCによってバルプロ酸ナトリウム含量について分析した。結果を透過総量(mg)で表した。3%(w/w)で局所適用されたVPAは、時間依存様態で皮膚に透過する。したがって、VPAは、局所適用される場合には、有効にヒト皮膚に透過することが可能である。
【0167】
実施例6
VPAは、ヒト不死化ケラチン生成細胞(HaCaT細胞)におけるヒストンH4アセチル化を濃度依存様態で誘発する(図7)。
【0168】
方法:
ウェスタンブロット:HaCaT細胞を処理24時間前に6ウェル培養皿へ接種した。培養物を指示濃度のVPAおよび/またはタザロテンで24時間処理した。全細胞抽出物を12%変性ポリアクリルアミドゲル上での変性SDSゲル電気泳動のためのRIPA緩衝液+プロテアーゼ阻害剤中の細胞溶解によって調製した。抗アセチル化H4抗体(クローンT25、特許出願EP02.021984.6号を参照)を使用するウェスタンブロット分析によってアセチル化ヒストンH4を検出し、同等のローディング対照として、抗アクチン抗体(シグマ(Sigma)、カタログ番号A5441)を使用してβ−アクチンの発現を確認した。
【0169】
結果:
図7において、上パネルは、指示通り処理されたHaCaT細胞からの全細胞ライセートの代表的な抗アセチル化ヒストンH4免疫ブロットを示す。下パネルは、ローディング対照としてのレプリカゲルの抗アクチン免疫ブロットを示す。細胞を0日目に6ウェルプレートにプレーティングした。薬剤を1日目に添加し、細胞を2日目にRIPA緩衝液+プロテアーゼ阻害剤で溶解した。全細胞ライセート40μgを12.5%SDS PAGEによって分離し、指示抗体で免疫ブロット法の対象にした。図7に示されているように、VPAは用量依存様態でヒストンH4高アセチル化を誘発する。
【0170】
実施例7
3%(w/w)で局所適用されたVPAは、マウス皮膚細胞(図8および9)およびヒト基底細胞癌(BCC)細胞(図10)の核におけるヒストンH4のアセチル化を誘発する。
【0171】
方法:
免疫組織化学:3%(w/w)で局所適用されるVPA(バルプロエート(Valproate))(製剤の詳細については実施例5を参照)またはプラセボ製剤を無胸腺ヌードマウスの皮膚に適用した。同様に、インフォームドコンセントを与えた後、3%(w/w)で適用されるVPA、またはプラセボ製剤を、外科的切除の16−20時間前に患者のBCC病変に局所適用した。3μmの凍結皮膚切片を室温下に10分間、アセトン中に固定した。抗アセチル化H4抗体(クローンT25、上記参照)での染色前に、ヤギ抗マウス免疫グロブリン(カペル(Cappel))(50mMトリス(Tris)緩衝食塩水、TBS中1/10000希釈)由来のF(ab’)2断片による30分間のプレインキュベーションによって組織をブロックした。TBSによる10分間の洗浄後、室温下に1時間、抗アセチル化H4抗体(クローンT25)(TBS中1/4000希釈)で切片をインキュベートした。次いで、試料をウサギ抗マウス血清でインキュベートし、APAAP(アルカリホスファターゼ抗アルカリホスファターゼ)免疫複合体で上記の通り処理した。陽性染色を、基質としてナフトール−AS−BIリン酸塩、かつ色原体としてニューフクシン(New Fuchsin)(ダコ(Dako))で赤色に発色させた。光学顕微鏡装置に接続したデジタルカメラで写真撮影した。
【0172】
結果:
皮膚生検を切除直後に液体窒素中で凍結させた。次いで、免疫組織化学を方法で記載した通り3μmの切片で行った。プラセボまたはVPAで局所治療した皮膚の生検を並行して処理し、同じ光度下にデジタルカメラに接続した光学顕微鏡で写真撮影した。
【0173】
図8および9に明らかに示されているように、局所VPA治療は、無胸腺ヌードマウスの皮膚細胞において、かつ―最も重要なことには―患者のBCC細胞および正常な皮膚細胞両者にヒストンH4アセチル化の明らかな増大を誘発した(図10)。これらのデータは、皮膚に局所適用されるVPAが皮膚細胞の高アセチル化を有効に誘発しうることを示す。
【0174】
実施例8
VPAは、0.1%(w/w)で適用されるタザロテンおよび3%(w/w)で適用されるVPAによる局所治療の前後6週間の代表的ば患者3例の写真に示されるように、表在性BCCの治療におけるタザロテンの効果を増強する(図11)。
【0175】
方法:
そのインフォームドコンセントを与えた後、平均年齢68歳の患者10例(男性6例、女性4例)を登録し、0.1%(w/w)のタザロテンおよび3%(w/w)で局所適用されるVPAで8週間1日1回、局所治療した。10個の病変、すなわち表在性BCC8個および色素性BCC2個を治療した。これらの病変のうち4個はすでにタザロテン単独で少なくとも16−20週間、前治療されており、この治療に対して完全に耐性であるように思われ、すなわち、タザロテン治療下に腫瘍直径の削減が確認されなかった。VPAをタザロテン適用の15−30分前に1日1回、局所適用した。病変を臨床的に評価し、2週間おきに写真を撮った。
【0176】
結果:
局所適用VPAおよびタザロテンによるBCC患者の治療は、6〜8週間の治療において全病変の50%を上回る後退(直径削減によって測定)をもたらした。これらの結果は、以前に未治療だった患者、および以前にタザロテンのみに暴露された患者の両方で確認された。興味深いことに、単独使用されたタザロテンは、BCCの数の後退(約50%)を誘発することがわかっているが、後退は通常ゆっくり(12−24週間)であり、一部の望ましくない副作用(疼痛や掻痒など)を伴う。しかし、VPAおよびタザロテンを組合せて使用する局所治療は明らかにタザロテンに対する反応を加速し、タザロテンによって引き起こされる望ましくない副作用の持続時間をも削減する。
【0177】
これらの結果により、VPAが他の化合物(タザロテンなど)の腫瘍細胞、特に皮膚の腫瘍の細胞に対する効果を増強しうるとともに―タザロテン耐性BCCの場合―VPAが腫瘍の耐性形態を感受性形態に変換しうることを示すことが確認される。全体的に、これらの結果により、皮膚過剰増殖性疾患の局所治療におけるVPAの有効性が確認される。
【0178】
実施例9
製剤例(軟膏)
白鉱油 20%
セチルアルコール 24%
セトマクロゴール1000 6%
バルプロ酸 3%
白ワセリン 100%に添加
【0179】
実施例10
製剤例 (ペースト)
酸化亜鉛 25%
でんぷん 25%
バルプロ酸 3%
白ワセリン 100%に添加
【0180】
実施例11
製剤例 (クリーム)
白ワセリン 25%
セチルアルコール 10%
トゥイーン(Tween)60 5%
グリセリン 10%
EDTA 0.2%
バルプロ酸ナトリウム 3%
再蒸留水 100%に添加
【0181】
実施例12
炎症性サイトカインなど免疫学的に関連するタンパク質の発現の調節。異なるHDAC阻害剤によるヒト不死化ケラチン生成細胞および末梢血リンパ球の処理は、結果として炎症性サイトカインの減少をもたらす(図12〜16)。
【0182】
方法:
ヒト不死化ケラチン生成細胞からの全RNAの単離
ヒト不死化ケラチン生成細胞(HaCaT細胞)を250万細胞/mlの密度で75cm2フラスコの中へ接種した。細胞を未処理で放置するか、または200nMトリコスタチンA(TSA)または5mMバルプロ酸(VPA)で4時間37℃下にプレインキュベートした後、リポ多糖類(LPS)(100ng/ml)で刺激した。37℃下に24時間後、細胞を溶解し、キアゲン(Qiagen)のRNeasyミニキットを使用して全RNAを単離した。
【0183】
末梢血単核細胞の単離および処理
単球およびマクロファージ枯渇末梢血単核細胞をFicoll−Hypaqueを使用する分離によって同意した成人から得た。末梢血単核細胞(PBMC)画分を洗浄し、9cmのペトリ皿に接種した。37℃下に2時間のインキュベーション後に単球、マクロファージ、およびB細胞の大部分を除去し、非接着細胞を回収し、175cm2のフラスコで2日間培養した。細胞を採取し、300万細胞/mlに調整した。500μlのアリコートを24ウェル平底プレートの各ウェルに移した。末梢血リンパ球(PBL)を指示通りさまざまな濃度のHDAC阻害剤で処理した。37℃下の2時間のインキュベーション後、細胞をホルボール12−ミリスチン酸13−アセテート(PMA)/イオノマイシン(Ion)で刺激し、または10μg/ml抗CD3mAb(OKT3)および2.5μg/ml抗CD28mAbとのT細胞受容体(TCR/CD3)複合体によって活性化した。37℃下に24時間後、上清を除去し、サイトカインアッセイ用に凍結した。細胞ペレットを溶解し、キアゲン(Qiagen)のRNeasyミニキットを使用して全RNAを単離した。
【0184】
RT−PCRおよび半定量PCR
全RNAの1マイクログラムを逆転写酵素およびオリゴ−dTプライマー(インビトロジェン(Invitrogen))使用する標準方法によってcDNAに転写した。半定量PCRのために、cDNA 2μlを特異的プライマーを使用するPCRによって増幅させた。PCR用のプライマーはMWGによって合成したが、それは以下の通りである。
【0185】
GAPDH: 5’−GGTGAAGGTCGGAGTCAACG−3’(配列番号:1)および
5’−CAAAGTTGTCATGGATGACC−3’(配列番号:2)、
IL−2: 5’−ATGTACAGGATGCAACTCCT−3’(配列番号:3)および
5’−TCAAGTTAGTGTTGAGATGA−3’(配列番号:4)、
IL−4: 5’−ATGGGTCTCACCTCCCAACT−3’(配列番号:5)および
5’−TCAGCTCGAACACTTTGAAT−3’(配列番号:6)、
IL−5: 5’−ATGAGGATGCTTCTGCATTTGAG−3’(配列番号:7)および
5’−TCCACTCGGTGTTCATTACACC−3’(配列番号:8)、
IL−6: 5’−ATGAACTCCTTCTCCACAAGCGCC−3’(配列番号:9)および
5’−CTACATTTGCCGAAGAGCCCTCAG−3’(配列番号:10)、
IL−8: 5’−ATGACTTCCAAGCTGGCCGTGGC−3’(配列番号:11)および
5’−TTATGAATTCTCAGCCCTCTTC−3’(配列番号:12)、
IL−10: 5’−TTGCCTGGTCCTCCTGACTG−3’(配列番号:13)および
5’−GATGTCTGGGTCTTGGTTCT−3’(配列番号:14)、
IL−12: 5’−ATGTGTCACCAGCAGTTGGTCATC−3’(配列番号:15)および
5’−CTATAGTAGCGGTCCTGGGC−3’(配列番号:16)、
TNF−α: 5’−ATGAGCACTGAAAGCATGATCCGG−3’(配列番号:17)および
5’−TCACAGGGCAATGATCCCAAAG−3’(配列番号:18)、
IFN−γ: 5’−ATGAAATATACAAGTTATATCTTGGCTTT−3’(配列番号:19)および
5’−TTACTGGGATGCTCTTCGAC−3’(配列番号:20)。
【0186】
IL−2およびTNF−αELISA
ELISAを行うために、処理および未処理PBLの上清を回収し、デュオセット(Duo Set)ELISA開発システム(R&Dシステムズ(R&D Systems))を使用し、メーカーによって記載されているようにIL−2およびTNF−αを測定した。
【0187】
ウェスタンブロット
全細胞抽出物をプロテアーゼ阻害剤を含む溶解緩衝液中で細胞溶解によって調製した。ライセートをSDSゲル電気泳動によって分離し、PVDF膜上に移した。アセチル化ヒストンH3およびH4を抗アセチル化H3抗体(アップステート(Upstate)、#06−942)、抗アセチル化H4抗体(クローンT25、特許出願EP02.021984.6号)、および抗−β−アクチン抗体を使用するウェスタンブロット分析によって検出した。同等のローディング用の対照としてβ−アクチン抗体を使用した。
【0188】
結果:
TSAおよび他のHDAC阻害剤による細胞の処理は、ヒストン高アセチル化および転写の調節をもたらす。したがって、本発明者らは、半定量RT−PCR分析によってサイトカインmRNAの発現レベル、およびELISAによってサイトカイン分泌に対するTSAおよび他のHDACの効果を調査した。
【0189】
ヒト不死化ケラチン生成細胞(HaCaT細胞)を、それぞれ、TSAまたはVPAの非存在下または存在下に24時間培養した。HDAC阻害剤で4時間のプレインキュベーション後、LPSを使用してサイトカイン産生を誘発した。サイトカインmRNA発現のレベルは半定量RT−PCRによって示されている(図12)。RT−PCR産物のアガロースゲル電気泳動は、未処理、ただし刺激対照と比べ、TSAおよびVPA処理細胞におけるTNF−αおよびIL−6mRNAのレベルにおける顕著な減少を示した(図12A)。これらの条件下、内部制御としてのGAPDH mRNAは影響を受けないままであった。ただし、LPS刺激によるIFN−γの誘導は適度に過ぎず、IFN−γのmRNA転写は実質的にTSAへの暴露によって影響されなかったが、VPAによって顕著に減少した。
【0190】
同様の結果は、末梢血リンパ球(PBL)を使用して得られた(図12B)。単離PBLをTSAおよびVPAで2時間プレインキュベートした後、37℃下に24時間PMA/Ionで刺激した。図12Bは、IL−4およびIL−6mRNA転写に対するTSAおよびVPAの効果を示す。トリコスタチンA(TSA)およびVPAは、IL−4のPMA/Ion介在刺激を顕著に減少させた。TSA処理後のIL−6mRNAでは適度の効果のみが確認されたが、VPAはIL−6mRNAを減少させ、刺激されていないサンプルで確認されるレベルに戻した。
【0191】
さらに、 図13AおよびBは、スベロイルアニリドヒドロキサム酸(SAHA)、G2M−701、G2M−702、およびG2M−707など他のHDAC阻害剤がサイトカイン発現を調節しうることを示す。
【0192】
図13に示されているように、さまざまなHDAC阻害剤は、IL−4およびIL−6mRNA転写の発現を減少させたが、GAPDHmRNA発現を修飾することはなかった。これらの条件下、IL−8mRNAは安定したままであり、PMA/IonによるT細胞活性化がこの遺伝子の発現を修飾することがないことを示した。
【0193】
相対的に、IL−2およびIFN−γ転写レベルに対するHDAC阻害剤の効果は、図14に示されているように、T細胞がCD3およびCD28抗体を使用してT細胞受容体複合体によって活性化された場合により顕著であった。PBLは、HDAC阻害剤TSA、SAHA、VPA、G2M−701、または抗炎症性ステロイドデキサメタゾン(Dex)でプレインキュベートされ、VPAおよびG2M−701は2つの異なる濃度で使用された。細胞は、24時間CD3およびCD28抗体を使用してT細胞受容体複合体(TCR/CD3)によって活性化された。図14AおよびBに示されているように、一部のサイトカインのmRNAは、わずかな相違で使用されたすべてのHDAC阻害剤によって顕著に減少した。図14Cは、アセチル化ヒストンH3およびアセチル化H4ならびに同等のローディングの対照としてのβ−アクチンに対する抗体を使用するウェスタンブロット分析を示し、HDAC阻害剤によるヒストン高アセチル化の有効な誘導を示した。
【0194】
半定量PCRの実験と一致する同様の結果が、図15に示されているようにELISAによるPBL培養上清における分泌IL−2およびTNF−αタンパク質レベルを分析することによって得られた。用量反応分析を行うために、VPA、G2M−701、G2M−702、およびG2M−707の濃度を上昇させてPBLを2時間、処理した後、CD3およびCD28mAbで24時間、37℃下に活性化した。上清を回収し、IL−2およびTNF−α分泌をELISAによって定量した。VPA、G2M−701、G2M−702、およびG2M−707によるPBLの治療は、結果として、IL−2およびTNF−α分泌の用量依存阻害をもたらした。阻害剤VPAの0.5mMおよび1mMが適度の効果のみを有したが、5mMはIL−2の分泌を顕著に減少した。
【0195】
これは、図16に示されているように、すでにVPAの1mMがIL−2およびTNF−α分泌における大幅な減少を示した、他の実験においてより顕著であった。IL−2およびTNF−α発現の阻害は、他のHDAC阻害剤でより有効であり、6μMのG2M−701、3μMのG2M−702、および1μMのG2M−707で最大であった(図15)。
【0196】
総合すれば、これらの結果は、VPA、G2M−701、G2M−702、およびG2M−707などHDAC阻害剤が、ヒトTリンパ球およびヒトケラチン生成細胞におけるサイトカイン発現のPMA/Ion(図12、13、および16)およびTCR/CD3(図14、15、および16)介在誘導を阻害することを示した。
【0197】
したがって、HDAC阻害剤は、細胞活性化に対する反応におけるサイトカイン発現を修飾する可能性を有する。それらは、免疫学的に重要な炎症性サイトカイン産生を無効にする一部のサイトカイン転写の発現をブロックすることができる。HDAC阻害剤によるサイトカイン分泌の劇的なダウンレギュレーションは、治療薬としてのその可能な使用を裏づける。
【0198】
実施例13
患者におけるHDAC阻害剤を使用する臨床治療データ
ヒストンデアセチラーゼクラスI酵素の選択的阻害剤として作用するVPAは、患者の細胞系および末梢血細胞におけるヒストン高アセチル化を誘発する。第I/II相臨床試験の範囲で静脈内VPAで治療した進行性悪性疾患を示す患者2例(患者#1および患者#2)から血液サンプルを採取した(図17、18、および19)。
【0199】
方法:
ウェスタンブロット
VPAで治療した患者からの末梢血細胞を、VPA治療の開始前、および開始後6時間、24時間、および48時間で得た(治療スケジュール、図17を参照)。全細胞抽出物をプロテアーゼ阻害剤を含むRIPA緩衝液中で細胞溶解によって調製した。ライセートをSDSゲル電気泳動によって分離し、PVDF膜上に移した。アセチル化ヒストンH3およびH4およびマーカー遺伝子HDAC−2を、抗アセチル化H3抗体(アップステート(Upstate)、#06−942)、抗アセチル化H4抗体(クローンT25、特許出願EP02.021984.6号)、および抗HDAC−2抗体(SCBT、SC−7899)を使用するウェスタンブロット分析によって検出した。同等のローディング対照PVDF膜をクーマシーで染色した(図18)。
【0200】
ELISA
VPA治療の開始前、開始後6時間、24時間、および48時間にVPAで治療した患者からの末梢血細胞を、100万細胞/mlの密度で24ウェル平底プレートへ接種した。細胞を刺激せずに放置し、またはCD3およびCD28抗体で刺激した。37℃下に24時間後、上清を回収し、IL−2およびTNF−αをELISA(R&Dシステムズ(R&D Systems))によって定量した(図19AおよびB)。
【0201】
RT−PCR
未刺激およびCD3/CD28刺激細胞からの全RNAをRNeasyミニキット(キアゲン(Qiagen))を使用して単離した。全RNAの1マイクログラムを逆転写酵素およびオリゴ−dTプライマー(インビトロジェン(Invitrogen))使用する標準方法によってcDNAに転写した。半定量PCRのために、cDNA 2μlを特異的プライマーを使用するPCRによって増幅させた(図19C)。
【0202】
結果:
末梢血細胞ライセートによるウェスタンブロット分析(図18)は、ヒストンH3およびH4高アセチル化、および治療血漿濃度を超える血清レベルでのマーカータンパク質HDAC−2のダウンレギュレーションの検出を示す。HDAC−2のヒストン高アセチル化およびダウンレギュレーションの誘導は、明らかにVPA治療の有効性を示したが、これはVPAが末梢血細胞におけるヒストン高アセチル化および標的遺伝子HDAC−2のレギュレーションを誘発する有効な治療血清濃度に達する患者において使用されうることを示す。また、本発明者らは、VPAが、ELISAによって分析されるように(図18A、およびB)培養上清におけるIL−2およびTNF−αなど炎症性サイトカインの発現を調節する証拠を示すが、これはCD3/CD28刺激細胞におけるIL−2およびTNF−α mRNA転写の減少と一致する(図18c)。さらに、これはVPA治療の24時間で開始するIL−4およびIFN−γのサイトカインmRNA発現を顕著に削減した。
【0203】
総合すれば、これらのデータは、VPAが、図17に示されている治療スケジュールによるHDAC阻害剤VPAで治療した患者におけるIL−2、TNF−α、IL−4、およびIFN−γなど免疫学的関連性遺伝子を有効に調節しうることを示す。
【0204】
したがって、免疫調節化合物として作用するVPAおよび他のHDAC阻害剤のこの新しい可能性は、病理学的に過剰活性の免疫細胞に結合される疾患の治療用の抗炎症薬としてこれらの化合物を使用する本発明を裏づける。
【図面の簡単な説明】
【0205】
【図1】VPAは、タザロテン(Taz)誘発細胞増殖阻害を増強する。 ヒト不死化ケラチン生成細胞(HaCaT細胞)で行われた増殖アッセイの結果を示し、単独で使用されたVPA、および単独で使用されたレチノイン酸誘導体タザロテンがHaCaT細胞増殖を阻害し、ヒストンデアセチラーゼ阻害剤VPAと組合せたタザロテンの使用は、単独で使用された各薬剤よりも細胞増殖の阻害でより有効であることを示す(NT:未処理、Taz:タザロテン、VPA1+Taz1:VPA 1mM+タザロテン 1μM、VPA1+Taz2:VPA 1mM+タザロテン 2μM)。
【図2】VPAは、レチノイン酸(RA)誘発細胞増殖阻害を増強する。 単独で使用されたVPAおよびRAがヒト不死化ケラチン生成細胞(HaCaT細胞)の増殖を阻害し、VPAがこれらの細胞において両方の薬剤が組合せて使用されるとレチノイン酸誘発細胞増殖阻害を増強することを示す(NT:未処理、RA:レチノイン酸、VPA1+RA1:VPA 1mM+レチノイン酸 1μM、VPA1+RA2:VPA 1mM+レチノイン酸 2μM)。
【図3】VPAは、5−フルオロウラシル(5−FU)誘発細胞増殖阻害を増強する。 単独のVPAおよび5−FUはHaCaT細胞の増殖を阻害し、5−FU(指示濃度で使用)+VPAの組合せは、単独で使用された各薬剤よりもHaCaT細胞増殖の阻害でより有効であることを示す(NT:未処理、5−FU:5−フルオロウラシル、VPAは1mMの濃度で5−FUと組合せて使用される)。
【図4】VPAは、ヒトケラチン生成細胞の細胞周期停止を誘発する。 VPAだけがヒト不死化ケラチン生成細胞(HaCaT細胞)における細胞周期停止を誘発することを示す。
【図5】VPAは、タザロテン誘発アポトーシスを増強する。 単独で使用されたタザロテンおよびVPAがHaCaT細胞のアポトーシスを誘発し、タザロテン+VPAの組合せが、単独で使用された各薬剤よりもこれらの細胞におけるアポトーシスの誘発でより有効であることを示す(NT:未処理、Taz:タザロテン、VPA1+Taz1:VPA 1mM+タザロテン 1μM、VPA1+Taz2:VPA 1mM+タザロテン 2μM)。
【図6】3%(w/w)で局所適用されるVPAは、ヒト皮膚に透過する。 3%(w/w)で局所適用されたVPAによるヒト皮膚の透過の時間依存様態を示す。
【図7】VPAは、ケラチン生成細胞におけるヒストンH4アセチル化を誘発する。 用量依存様態でHaCaT細胞におけるVPA誘発ヒストンH4アセチル化を示す。
【図8】3%(w/w)で局所適用されるVPAは、マウスにおける皮膚細胞アセチル化を誘発する。 3%(w/w)で局所適用されたVPAで処理されたマウスの皮膚細胞におけるヒストンH4アセチル化を示す。
【図9】3%(w/w)で局所適用されるVPAは、マウスにおける皮膚細胞アセチル化を誘発する。 図8における表示と比べて高い倍率における3%(w/w)でのVPAの局所適用により誘発されたマウスの皮膚細胞におけるヒストンH4アセチル化を示す。
【図10】3%(w/w)で局所適用されるVPAは、BCC核のアセチル化を誘発する。 3%(w/w)でのVPAの局所適用後のヒト基底細胞癌(BCC)におけるヒストンH4のアセチル化の誘導を示す。
【図11】3%(w/w)で局所適用されるVPAは、表在性BCCの治療におけるタザロテンの効果を増強する。 治療前、および0.1%(w/w)で適用されたタザロテンおよび3%(w/w)で適用されたVPAによる併用局所治療の6週間後の3人の代表的な患者の例を示す。
【図12】ヒトケラチン生成細胞および末梢血リンパ球におけるTSAおよびVPAによる炎症性サイトカインの調節。 LPSまたはPMA/Ionによる刺激後のHDAC阻害剤による炎症性サイトカインなど免疫学的に関連する遺伝子のmRNAの発現調節を示す(実施例12)。
【図13】異なるHDAC阻害剤による炎症性サイトカインの調節。 PMA/Ionによる刺激後のHDAC阻害剤による炎症性サイトカインなど免疫学的に関連する遺伝子のmRNAの発現調節を示す(実施例12)。
【図14】末梢血リンパ球におけるHDAC阻害剤による炎症性サイトカインの調節。 CD3/CD28刺激後のHDAC阻害剤による炎症性サイトカインなど免疫学的に関連する遺伝子のmRNAの発現調節を示す(実施例12)。
【図15】末梢血リンパ球におけるHDAC阻害剤によるIL−2およびTNF−α発現の調節。 CD3/CD28刺激後のHDAC阻害剤による炎症性サイトカインなど免疫学的に関連する遺伝子のタンパク質の発現調節を示す(実施例12)。
【図16】PMA/IONおよびCD3/CD28mABで刺激した末梢血リンパ球におけるHDAC阻害剤によるIL−2およびTNF−α発現の調節。 HDAC阻害剤による炎症性サイトカインなど免疫学的に関連性の遺伝子のタンパク質発現調節を示す(実施例12)。
【図17】第I/II相試験からの患者のVPA治療スケジュール。 癌患者におけるHDAC阻害剤VPAを用いた臨床治療スケジュール(実施例13)を示す。
【図18】VPAは、第I/II相試験からの患者の末梢血におけるヒストン高アセチル化およびマーカー遺伝子の制御を誘発する。 図17に示されている治療スケジュールによるHDAC阻害剤VPAにより治療された患者の末梢血におけるHDAC−2タンパク質のヒストンアセチル化およびダウンレギュレーションの有効な誘導を示す(実施例13)。
【図19】第I/II相試験における患者からのVPAによる炎症性サイトカインの調節。 CD3/CD28刺激後の図17に示されている治療スケジュールによる患者におけるHDAC阻害剤VPAを使用することによって炎症性サイトカインなど免疫学的に関連する遺伝子の有効なmRNAおよびタンパク質の発現調節を示す(実施例13)。
【特許請求の範囲】
【請求項1】
皮膚疾患の予防または治療のための局所医薬組成物であって、
(i)VPAおよびその医薬的に許容される塩、VPAの誘導体およびその医薬的に許容される塩から成る群より選択される少なくとも約0.1%の活性剤と、
(ii)皮膚科学的に許容される担体と
を含んでなる局所医薬組成物。
【請求項2】
前記活性剤の濃度が前記組成物の約0.1%〜約6%である、請求項1に記載の局所組成物。
【請求項3】
レチノイン酸またはその誘導体をさらに含んでなる、請求項1または2に記載の局所組成物。
【請求項4】
前記レチノイン酸またはその誘導体の濃度が前記組成物の約0.01%〜約1%である、請求項3に記載の局所組成物。
【請求項5】
前記レチノイン酸の誘導体が、9−シスレチノイン酸、トランス−レチノイン酸、全トランスレチノイン酸、およびタザロテンから成る群より選択される、請求項3または4に記載の局所組成物。
【請求項6】
化学療法薬をさらに含んでなる、請求項1〜5のいずれか一項に記載の局所組成物。
【請求項7】
前記化学療法薬が5−フルオロウラシルである、請求項6に記載の局所組成物。
【請求項8】
前記化学療法薬の濃度が組成物の約0.1%〜約10%である、請求項6または7に記載の局所組成物。
【請求項9】
前記皮膚科学的に許容される担体が、クリーム、軟膏、ペースト、およびゲルから成る群より選択される、請求項1〜8のいずれか一項に記載の局所組成物。
【請求項10】
皮膚疾患の予防または治療のための医薬製造用のVPAまたはその誘導体の使用であって、VPAおよびその医薬的に許容される塩、VPAの誘導体およびその医薬的に許容される塩から成る群より選択される活性剤を含んでなる医薬組成物が個人の皮膚に局所適用される使用。
【請求項11】
前記治療が、約1cm2の病変当り0.5〜10mg/日の投与を含んでなる、請求項10に記載の使用。
【請求項12】
前記皮膚疾患が、タンパク質の高アセチル化の誘導が患者に対する有利な治療効果を有するヒト皮膚の疾患である、請求項10または11に記載の使用。
【請求項13】
前記皮膚疾患が、皮膚腫瘍、前腫瘍性皮膚疾患、皮膚および/または粘膜の炎症および光老化から成る群より選択される、請求項10〜12のいずれか一項に記載の使用。
【請求項14】
前記皮膚疾患が、基底細胞癌、扁平上皮細胞癌、角化棘細胞腫、ボーエン病および皮膚T細胞リンパ腫から成る群より選択される、請求項13に記載の使用。
【請求項15】
前記皮膚疾患が、前腫瘍性皮膚疾患光線性角化症である、請求項13に記載の使用。
【請求項16】
前記皮膚疾患が、乾癬、魚鱗癬、および座瘡から成る群より選択される皮膚および/または粘膜の炎症である、請求項13に記載の使用。
【請求項17】
前記活性剤および別の活性剤が、同じ製剤または別個の製剤で局所適用される、請求項10〜16のいずれか一項に記載の使用。
【請求項18】
VPAと異なるヒストンデアセチラーゼの別の阻害剤が使用される、請求項10〜17のいずれか一項に記載の使用。
【請求項19】
前記VPAと異なるヒストンデアセチラーゼの阻害剤が、NVP−LAQ824、トリコスタチンA(TSA)、スベロイルアニリドヒドロキサム酸、CBHA、ピロキサミド(Pyroxamide)、スクリプタイド(Scriptaid)、CI−994、CG−1521、クラミドシン(Chlamydocin)、ビアリールヒドロキサム酸塩、例えば、A−161906、二環式アリール−N−ヒドロキシカルボキサミド、PXD−101、スルホンアミドヒドロキサム酸、TPX−HA類似体(CHAP)、オキサムフラチン(Oxamflatin)、トラポキシン(Trapoxin)、デプデシン(Depudecin)、アピジシン(Apidicin)などヒドロキサム酸誘導体、MS−27−27、ピロキサミドおよびその誘導体などベンズアミド、酪酸、およびその誘導体、例えば、ピバネックス(Pivanex)(ピバロイルオキシメチルブチレート)など短鎖脂肪酸、トラポキシンA、デプシペプチド(FK−228)および関連ペプチド化合物、タセジナリン(Tacedinaline)、MG2856など環状テトラペプチド、およびHDACクラスIII阻害剤、またはSIRT阻害剤から成る群より選択される、請求項18に記載の使用。
【請求項20】
VPAもしくはその誘導体、または請求項18または19において規定されたVPAと異なるヒストンデアセラーゼの別の阻害剤が、VPAもしくはその誘導体またはヒストンデアセラーゼの別の阻害剤のみの局所適用によって、皮膚腫瘍;基底細胞癌;扁平上皮細胞癌;角化棘細胞腫;ボーエン病;皮膚T細胞リンパ腫;光線性角化症;乾癬;魚鱗癬;座瘡;他の炎症性皮膚疾患を含む腫瘍性および非腫瘍性皮膚疾患における経口適用レチノイド活性を増強する、請求項10〜19のいずれか一項に記載の使用。
【請求項21】
VPAもしくはその誘導体、または請求項18または19において規定されたVPAと異なるヒストンデアセラーゼの別の阻害剤が、VPAもしくはその誘導体またはヒストンデアセラーゼの別の阻害剤のみの局所適用によって、皮膚腫瘍;基底細胞癌;扁平上皮細胞癌;角化棘細胞腫;ボーエン病;皮膚T細胞リンパ腫;光線性角化症;乾癬;魚鱗癬;座瘡;他の炎症性皮膚疾患を含む腫瘍性および非腫瘍性皮膚疾患におけるタカルシトールおよびカルシポトリオール活性など経口適用ビタミンD誘導体を増強する、請求項10〜20のいずれか一項に記載の使用。
【請求項22】
前記治療が、請求項1〜9のいずれか一項に記載の局所医薬組成物の投与を含んでなる、請求項10〜21のいずれか一項に記載の使用。
【請求項23】
前記VPAの誘導体が式I
【化1】
[式中、R1およびR2は独立して、場合により1個もしくは何個かのヘテロ原子を含んで成り、かつ置換されうる直鎖または分岐、飽和または不飽和、脂肪族C3-25炭化水素鎖であり、R3はヒドロキシル、ハロゲン、アルコキシ、または場合によりアルキル化アミノ基、またはその医薬的に許容される塩である]の化合物である、請求項1〜22のいずれか一項に記載の組成物または使用。
【請求項24】
R1およびR2が独立して、場合により二重または三重結合を含んでなる直鎖または分岐C3-25炭化水素鎖である、請求項1〜23のいずれか一項に記載の組成物または使用。
【請求項25】
前記VPA誘導体が、4−yn−VPAまたはその医薬的に許容される塩である、請求項1〜24のいずれか一項に記載の組成物または使用。
【請求項1】
皮膚疾患の予防または治療のための局所医薬組成物であって、
(i)VPAおよびその医薬的に許容される塩、VPAの誘導体およびその医薬的に許容される塩から成る群より選択される少なくとも約0.1%の活性剤と、
(ii)皮膚科学的に許容される担体と
を含んでなる局所医薬組成物。
【請求項2】
前記活性剤の濃度が前記組成物の約0.1%〜約6%である、請求項1に記載の局所組成物。
【請求項3】
レチノイン酸またはその誘導体をさらに含んでなる、請求項1または2に記載の局所組成物。
【請求項4】
前記レチノイン酸またはその誘導体の濃度が前記組成物の約0.01%〜約1%である、請求項3に記載の局所組成物。
【請求項5】
前記レチノイン酸の誘導体が、9−シスレチノイン酸、トランス−レチノイン酸、全トランスレチノイン酸、およびタザロテンから成る群より選択される、請求項3または4に記載の局所組成物。
【請求項6】
化学療法薬をさらに含んでなる、請求項1〜5のいずれか一項に記載の局所組成物。
【請求項7】
前記化学療法薬が5−フルオロウラシルである、請求項6に記載の局所組成物。
【請求項8】
前記化学療法薬の濃度が組成物の約0.1%〜約10%である、請求項6または7に記載の局所組成物。
【請求項9】
前記皮膚科学的に許容される担体が、クリーム、軟膏、ペースト、およびゲルから成る群より選択される、請求項1〜8のいずれか一項に記載の局所組成物。
【請求項10】
皮膚疾患の予防または治療のための医薬製造用のVPAまたはその誘導体の使用であって、VPAおよびその医薬的に許容される塩、VPAの誘導体およびその医薬的に許容される塩から成る群より選択される活性剤を含んでなる医薬組成物が個人の皮膚に局所適用される使用。
【請求項11】
前記治療が、約1cm2の病変当り0.5〜10mg/日の投与を含んでなる、請求項10に記載の使用。
【請求項12】
前記皮膚疾患が、タンパク質の高アセチル化の誘導が患者に対する有利な治療効果を有するヒト皮膚の疾患である、請求項10または11に記載の使用。
【請求項13】
前記皮膚疾患が、皮膚腫瘍、前腫瘍性皮膚疾患、皮膚および/または粘膜の炎症および光老化から成る群より選択される、請求項10〜12のいずれか一項に記載の使用。
【請求項14】
前記皮膚疾患が、基底細胞癌、扁平上皮細胞癌、角化棘細胞腫、ボーエン病および皮膚T細胞リンパ腫から成る群より選択される、請求項13に記載の使用。
【請求項15】
前記皮膚疾患が、前腫瘍性皮膚疾患光線性角化症である、請求項13に記載の使用。
【請求項16】
前記皮膚疾患が、乾癬、魚鱗癬、および座瘡から成る群より選択される皮膚および/または粘膜の炎症である、請求項13に記載の使用。
【請求項17】
前記活性剤および別の活性剤が、同じ製剤または別個の製剤で局所適用される、請求項10〜16のいずれか一項に記載の使用。
【請求項18】
VPAと異なるヒストンデアセチラーゼの別の阻害剤が使用される、請求項10〜17のいずれか一項に記載の使用。
【請求項19】
前記VPAと異なるヒストンデアセチラーゼの阻害剤が、NVP−LAQ824、トリコスタチンA(TSA)、スベロイルアニリドヒドロキサム酸、CBHA、ピロキサミド(Pyroxamide)、スクリプタイド(Scriptaid)、CI−994、CG−1521、クラミドシン(Chlamydocin)、ビアリールヒドロキサム酸塩、例えば、A−161906、二環式アリール−N−ヒドロキシカルボキサミド、PXD−101、スルホンアミドヒドロキサム酸、TPX−HA類似体(CHAP)、オキサムフラチン(Oxamflatin)、トラポキシン(Trapoxin)、デプデシン(Depudecin)、アピジシン(Apidicin)などヒドロキサム酸誘導体、MS−27−27、ピロキサミドおよびその誘導体などベンズアミド、酪酸、およびその誘導体、例えば、ピバネックス(Pivanex)(ピバロイルオキシメチルブチレート)など短鎖脂肪酸、トラポキシンA、デプシペプチド(FK−228)および関連ペプチド化合物、タセジナリン(Tacedinaline)、MG2856など環状テトラペプチド、およびHDACクラスIII阻害剤、またはSIRT阻害剤から成る群より選択される、請求項18に記載の使用。
【請求項20】
VPAもしくはその誘導体、または請求項18または19において規定されたVPAと異なるヒストンデアセラーゼの別の阻害剤が、VPAもしくはその誘導体またはヒストンデアセラーゼの別の阻害剤のみの局所適用によって、皮膚腫瘍;基底細胞癌;扁平上皮細胞癌;角化棘細胞腫;ボーエン病;皮膚T細胞リンパ腫;光線性角化症;乾癬;魚鱗癬;座瘡;他の炎症性皮膚疾患を含む腫瘍性および非腫瘍性皮膚疾患における経口適用レチノイド活性を増強する、請求項10〜19のいずれか一項に記載の使用。
【請求項21】
VPAもしくはその誘導体、または請求項18または19において規定されたVPAと異なるヒストンデアセラーゼの別の阻害剤が、VPAもしくはその誘導体またはヒストンデアセラーゼの別の阻害剤のみの局所適用によって、皮膚腫瘍;基底細胞癌;扁平上皮細胞癌;角化棘細胞腫;ボーエン病;皮膚T細胞リンパ腫;光線性角化症;乾癬;魚鱗癬;座瘡;他の炎症性皮膚疾患を含む腫瘍性および非腫瘍性皮膚疾患におけるタカルシトールおよびカルシポトリオール活性など経口適用ビタミンD誘導体を増強する、請求項10〜20のいずれか一項に記載の使用。
【請求項22】
前記治療が、請求項1〜9のいずれか一項に記載の局所医薬組成物の投与を含んでなる、請求項10〜21のいずれか一項に記載の使用。
【請求項23】
前記VPAの誘導体が式I
【化1】
[式中、R1およびR2は独立して、場合により1個もしくは何個かのヘテロ原子を含んで成り、かつ置換されうる直鎖または分岐、飽和または不飽和、脂肪族C3-25炭化水素鎖であり、R3はヒドロキシル、ハロゲン、アルコキシ、または場合によりアルキル化アミノ基、またはその医薬的に許容される塩である]の化合物である、請求項1〜22のいずれか一項に記載の組成物または使用。
【請求項24】
R1およびR2が独立して、場合により二重または三重結合を含んでなる直鎖または分岐C3-25炭化水素鎖である、請求項1〜23のいずれか一項に記載の組成物または使用。
【請求項25】
前記VPA誘導体が、4−yn−VPAまたはその医薬的に許容される塩である、請求項1〜24のいずれか一項に記載の組成物または使用。
【図1】

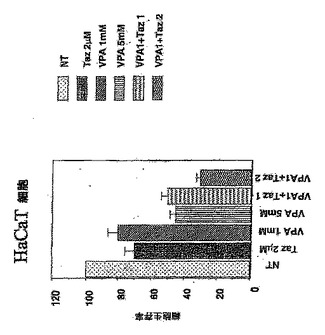
【図2】
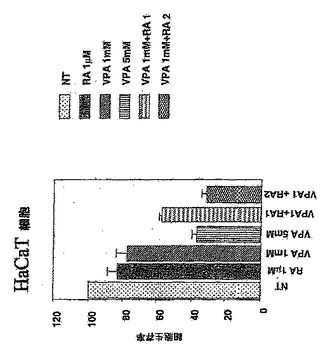
【図3】

【図4】

【図5】

【図6】

【図7】
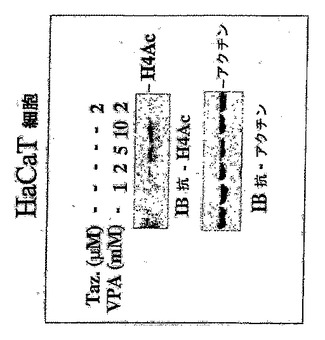
【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】
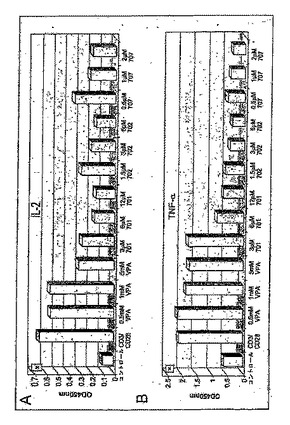
【図16】

【図17】

【図18】

【図19】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公表番号】特表2008−529964(P2008−529964A)
【公表日】平成20年8月7日(2008.8.7)
【国際特許分類】
【出願番号】特願2006−516036(P2006−516036)
【出願日】平成16年6月23日(2004.6.23)
【国際出願番号】PCT/EP2004/006789
【国際公開番号】WO2005/000289
【国際公開日】平成17年1月6日(2005.1.6)
【出願人】(505474016)トポターゲット ジャーマニー アーゲー (2)
【Fターム(参考)】
【公表日】平成20年8月7日(2008.8.7)
【国際特許分類】
【出願日】平成16年6月23日(2004.6.23)
【国際出願番号】PCT/EP2004/006789
【国際公開番号】WO2005/000289
【国際公開日】平成17年1月6日(2005.1.6)
【出願人】(505474016)トポターゲット ジャーマニー アーゲー (2)
【Fターム(参考)】
[ Back to top ]