
Fターム[4C072UU01]の内容
N及び(O又はS)縮合複素環 (24,645) | 化合物の用途 (2,401) | 医薬 (1,977)
Fターム[4C072UU01]に分類される特許
1,941 - 1,960 / 1,977
キメラGABAB受容体
本発明は、少なくとも1つのGABABR1aサブユニットおよび少なくとも1つのGABABR2サブユニットを含んでなり、該GABAB受容体が1つの高親和性アゴニスト結合部位および1つの低親和性アゴニスト結合部位を有することを特徴とする単離されたGABAB受容体タンパク質を提供する。特に、受託番号LMBP 6046CBで2003年8月22日にCHO−K1 h−GABA−b R1a/R2クローンとしてBelgian Coordinated Collections of Microorganisms(BCCM)に寄託されたhGABABR1a/GABABR2 CHO細胞系により発現される単離された組み換えGABAB受容体タンパク質。従って、受託番号LMBP 6046CBで2003年8月22日にCHO−K1 h−GABA−b R1a/R2クローンとしてBelgian Coordinated Collections of Microorganisms(BCCM)に寄託されたhGABABR1a/GABABR2 CHO細胞系を提供することが本発明の目的である。本発明はまた、機能もしくは結合アッセイを用いてGABAB受容体アゴニストを同定する方法における上記の細胞系の使用も提供する。特に、例えば3H−GABAもしくは3H−バクロフェンのような放射性標識アゴニストの使用を含んでなる放射性リガンド結合アッセイにおける。特定の態様として、本発明は機能もしくは結合アッセイを用いて高親和性GABAB受容体アゴニストを同定する方法における上記のGABAB受容体の使用を提供する。特に、例えば3H−GABAもしくは3H−バクロフェンのような放射性標識アゴニストの使用を含んでなる放射性リガンド結合アッセイにおける。あるいはまた、上記の結合アッセイは、細胞抽出物、特に上記の細胞の細胞膜調製物上で行われる。 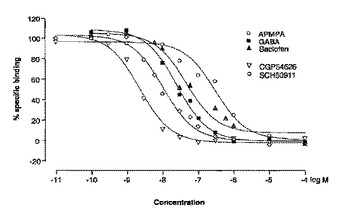 (もっと読む)
(もっと読む)
ククルビットウリルの製造方法
本発明は、ククルビットウリルの製造方法に関する。この方法は、ククルビットウリルを形成するために酸の存在下において、グリコールウリル、グリコールウリル類縁体及び/又はグリコールウリルもしくはグリコールウリル類縁体のオリゴマーから選択される1つ以上の化合物と、2から11の結合されたグリコールウリル又はグリコールウリル類縁体からなるオリゴマーを反応させることを含む。該方法は、ククルビットウリルにおける特定の位置に特定の置換されたユニットを有する可変の置換ククルビットウリルを製造するために使用し得る。本発明はまた、本発明の方法により製造されるククルビットウリルを提供する。本発明はさらに、新規なククルビットウリルを提供する。 (もっと読む)
CRF1受容体リガンドである、ヘテロアリール縮合ピリジン類、ピラジン類及びピリミジン類
CRF1受容体の選択的調節剤として作用する、置換ヘテロアリール縮合ピリジン、ピラジン及びピリミジン化合物を提供する。これら化合物は、多くのCNS及び末梢障害、特にストレス、不安神経症、うつ病、心臓血管障害、及び摂食障害の治療に有用である。このような障害の治療方法、更に包装された医薬組成物も提供する。本発明の化合物は、CRF受容体の局在化のためのプローブとして、また、CRF受容体結合試験の標準としても有用である。受容体局在化の研究に当該化合物を使用する方法も提供する。 (もっと読む)
THIPの製造方法
本発明は睡眠障害の治療に有用なガボクサドール(THIP)の新規製造方法に関する。特に、式(8b)の化合物またはその塩であって、式中、Rが場合により1つまたはそれ以上のC1-12アルキル、C1-12アルコキシまたはアリールで置換されるC1-12アルキル、C2-12アルケニル、C3-8シクロアルキル、C3-8シクロアルケニル、アシルまたはアリールであり、U’ がNH、またはR1がHまたはRであるCHR1であり、WがO、S、またはR4がHまたはRであるNR4である前記式(8b)の化合物またはその塩を、酸、典型的には無機酸と反応させて、酸付加塩としてTHIPを得ることを含む、THIPの製造方法に関する。
本発明は、またいくつかの中間体に関する。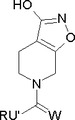
(8b)
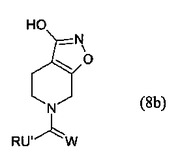 (もっと読む)
(もっと読む)
置換8−ヘテロアリールキサンチン
本発明は、A2Bアデノシン受容体(AR)の選択性アンタゴニストである化合物および医薬組成物を提供する。これらの化合物および組成物は医薬として有用である。 (もっと読む)
ピリミドチオフェン化合物
式(I):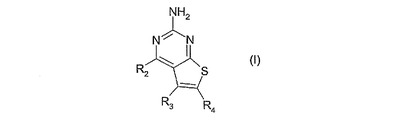
[式中、
R2は、式-(Ar1)m-(Alk1)p-(Z)r-(Alk2)s-Q(ここで、Ar1は任意に置換されていてもよいアリールまたはヘテロアリール基であり、Alk1およびAlk2は任意に置換されていてもよい2価のC1-C3アルキレンまたはC2-C3アルケニレン基であり、m、p、rおよびsは独立して0または1であり、Zは-O-、-S-、-(C=O)-、-(C=S)-、-SO2-、-C(=O)O-、-C(=O)NRA-、-C(=S)NRA-、-SO2NRA-、-NRAC(=O)-、-NRASO2-または-NRA-(ここで、RAは水素またはC1-C6アルキルである)であり、そして
Qは水素または任意に置換されていてもよい炭素環式もしくはヘテロ環式基である)の基であり;
R3は水素、任意の置換基、または任意に置換されていてもよい(C1-C6)アルキル、アリールもしくはヘテロアリール基であり;そして
R4はカルボキシエステル、カルボキシアミドまたはスルホンアミド基である]
の化合物はインビトロまたはインビボでのHSP90の阻害剤であり、それらは特に癌の治療に役立つ。 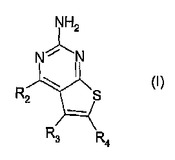 (もっと読む)
(もっと読む)
粘膜に関連した症状の処置に関する方法
IRMを粘膜表面に間欠的に適用することによるIRMの断続的送達を使用して、刺激副作用をかなり低減させつつ、サイトカイン誘導による治療レベルおよび治療持続時間を達成することが可能である。 (もっと読む)
IV型ホスホジエステラーゼの阻害剤
【解決課題】式Iのイソオキサゾリン誘導体であって、IV型ホスホジエステラーゼ(PDE)の選択的阻害剤として使用することができる誘導体に関する。
【解決手段】本明細書中で開示される化合物は、患者、特にヒトの、エイズ、喘息、関節炎、気管支炎、慢性閉塞性肺疾患(COPD)、乾癬、アレルギー性鼻炎、ショック、アトピー性皮膚炎、クローン病、成人呼吸促迫症候群(ARDS)、エオシン好性肉芽腫、アレルギー性結膜炎、骨関節炎、潰瘍性大腸炎および他の炎症性疾患の処置に有用である。また、開示されている化合物の製造方法、ならびにその医薬組成物、およびIV型ホスホジエステラーゼ(PDE)阻害剤とその利用に関する。
式I 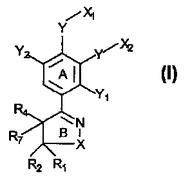 (もっと読む)
(もっと読む)
胃腸管運動障害を処置するために有用な組成物
本発明は、第1の量の5−HT3受容体アゴニスト活性またはその薬学的に許容され得る塩、水和物、もしくは溶媒和物を有する化合物、および第2の量の少なくとも1つの胃酸抑制剤(例えば、プロトンポンプ阻害薬、H2受容体アンタゴニストおよびその薬学的に許容され得る塩、水和物または溶媒和物、またはアシッドポンプアンタゴニストもしくはその薬学的に許容され得る塩、水和物、もしくは溶媒和物)を、被検体に同時投与することを含む、処置の必要な被検体における胃腸管運動障害を処置する方法に関し、ここで第1または第2の量は共に治療有効量を含む。特に、該方法はGERDを処置するためであり、夜間GERDを含む。本発明は更に、5−HT3受容体アゴニスト活性、またはその薬学的に許容され得る塩、水和物、もしくは溶媒和物を有する化合物の治療有効量を被験体に投与することを含む、処置の必要な被験体における夜間GERDを処置する方法に関する。本発明は更に、それの必要な被験体における増加する食道の運動の方法に関する。増加する食道の運動の方法は、5−HT3受容体アゴニスト活性、またはその薬学的に許容され得る塩、水和物、もしくは溶媒和物を有する化合物を投与することにより得られ得る。同時投与はまた、食道の運動を増加させるために使用され得る。 (もっと読む)
統合失調症の治療のための[1,8]ナフチリジン−2−オンおよび関連化合物
本発明は、G、A、Z、Q、X、YおよびR1およびR2が本明細書におけるように定義される式(1)の化合物、それらを含有する薬剤組成物ならびに中枢神経系およびその他の疾患の治療におけるそれらの使用に関する。 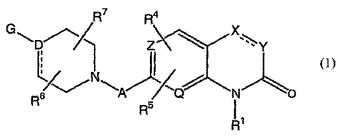 (もっと読む)
(もっと読む)
免疫刺激性の組み合わせおよび治療
本発明は、免疫刺激性の組み合わせおよび方法を提供する。一般に、この免疫刺激性の組み合わせは、IRM化合物の局所製剤と、医薬組成物と、を含む。一般に、この方法は、(a)IRM化合物の局所製剤と、(b)医薬組成物と、を対象の投与部位に投与することを含む。  (もっと読む)
(もっと読む)
近赤外線造影剤に適する3H−フェノキサジン誘導体、その製造法および使用
本発明は、遊離塩基形または酸付加塩形の、式I
【化1】
〔式中、XおよびYは、XおよびYが同時にCHまたはCH2ではないとの条件下で、CH、CH2または2価もしくは3価ヘテロ原子であり;mおよびoは、互いに独立して0または1である。ただし、mが0であるとき、Yと隣接C原子の間の点線は結合であり、かつYはCHまたは3価ヘテロ原子であり、mが1であるとき、Yと隣接C原子の間の点線は存在せず、かつYはCH2または2価ヘテロ原子であり、oが0であるとき、Xと隣接C原子の間の点線は結合であり、かつXはCHまたは3価ヘテロ原子であり、oが1であるとき、Xと隣接C原子の間の点線は存在せず、かつXはCH2または2価ヘテロ原子である;Aは(CR3R4)pであり、そしてQは(CR9R10)nであり;nおよびpは、互いに独立して0または1であり;R6、R7、R13、およびR14は、互いに独立して水素、ハロゲン、(C1−4)アルキル、(C1−4)アルキルSO2、SO3H、カルボキシ、(C1−4)アルコキシカルボニル、(C1−4)アルコキシ、OHまたはNR15R16であり;R1、R2、R3、R4、R9、R10、R11およびR12は、互いに独立して水素、(C1−4)アルキル、カルボキシ、(C1−4)アルコキシカルボニルまたは(C1−4)アルコキシであるか、または、XがCHもしくはCH2であるとき、R1およびR2はまたOHもしくはNR15R16であり得るか、またはYがCHもしくはCH2であるとき、R11、R12はまたOHもしくはNR15R16あり得て;R5、R8、R15およびR16は、互いに独立して水素、(C1−4)アルキル、(C1−4)アルコキシ、R17O−C(O)−(C1−4)アルキルまたは(反応性基)−(C1−4)アルキルであり;そしてR17は水素または(C1−4)アルキルである。〕の化合物を提供する。
 (もっと読む)
(もっと読む)
2,4−ピリミジンジアミン化合物による自己免疫疾患の治療または予防方法
本発明は、2,4−ピリミジンジアミン化合物によって、自己免疫疾患を治療あるいは予防する方法、およびこのような疾病にまつわる症状を治療、予防、あるいは改善する方法を提供する。当該化合物によって治療または予防される自己免疫疾患の具体例として、関節リウマチおよび・またはそれにまつわる症状、全身性エリトマトーデスおよび・またはそれにまつわる症状、また多発性硬化症および/またはそれにまつわる症状が含まれる。 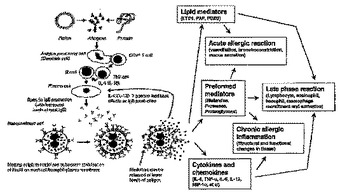 (もっと読む)
(もっと読む)
新生物疾患、炎症および免疫障害の処置に有用な2,4−ピリミジンジアミン
式I
【化1】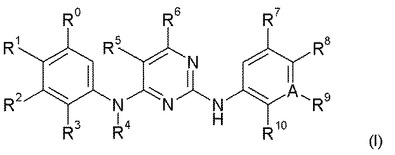
〔式中、RはC6−10アリール、C5−10ヘテロアリール、C3−12シクロアルキルおよびC3−10ヘテロシクロアルキルから選択され;R0−R6は本明細書で記載の通りである。〕のピリミジン誘導体;およびFAKおよび/またはALKおよび/またはZAP−70および/またはIGF−IRの阻害に応答する疾患の処置用医薬の製造のためのそれらの使用。
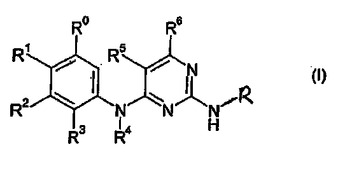 (もっと読む)
(もっと読む)
CCI−779の位置選択的合成
インドール−4スルホンアミド誘導体、それらの調製および5−HT−6調節因子としてのそれらの使用
本発明は、一般式(Ia、Ib、Ic)の新規スルホンアミド誘導体であって、任意で、それらの立体異性体、好ましくはエナンチオマーもしくはジアステレオマー、それらのラセミ化合物の形態であるか、またはいずれかの混合比にあるそれらの立体異性体、好ましくはエナンチオマーもしくはジアステレオマーの少なくとも2つの混合物の形態である誘導体、またはそれらの塩、好ましくは対応する生理的に許容される塩、または対応する溶媒和物に関するともに、それらを調製する方法、ヒトおよび/または獣医学的治療用の薬剤としてのそれらの使用、ならびにそれらを含有する医薬組成物に関する。
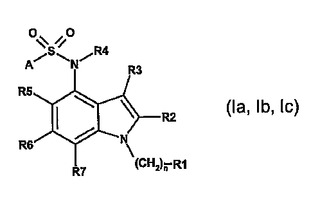 (もっと読む)
(もっと読む)
インドール−6スルホンアミド誘導体、その調製及び5−HT−6モジュレータとしてのその使用
本発明は、任意選択的にその立体異性体、好ましくはエナンチオマー又はジアステレオマーのうちの1つ、そのラセミ体の形の、あるいはその立体異性体、好ましくはエナンチオマー又はジアステレオマーのうちの少なくとも2つの、任意の混合比での混合物の形の、一般式(Ia、Ib、Ic)の新しいスルホンアミド誘導体、又はその塩、好ましくはその該当する生理学的に許容される塩、又はその該当する溶媒和物に;その調製のための工程に、ヒト及び/又は動物治療学における医薬品でのその利用に、及びそれを含有する医薬組成物に関するものである。
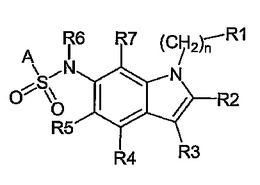 (もっと読む)
(もっと読む)
抗増殖剤としての2,4−ピリミジンジアミン化合物および使用
本発明は、抗増殖活性を有する2,4−ピリミジンジアミン化合物、これらの化合物を含有する組成物、およびこれらの化合物を使用して細胞増殖を阻止し増殖疾患(例えば、腫瘍形成癌)を治療する方法を提供する。他の局面では、本発明は、これらの2,4−ピリミジンジアミン化合物のプロドラッグを提供する。このようなプロドラッグは、それらのプロドラッグ形状で活性であり得るか、または使用する生理学的条件または他の条件下にて活性薬剤形状に変換されるまで、不活性であり得る。 (もっと読む)
インドール−5スルホンアミド誘導体、それらの調製及び5−HT−6モジュレータとしてのそれらの使用
本発明は、任意選択的にその立体異性体、好ましくはエナンチオマー又はジアステレオマーのうちの1つ、そのラセミ体の形の、あるいはその立体異性体、好ましくはエナンチオマー又はジアステレオマーのうちの少なくとも2つの、任意の混合比での混合物の形の、一般式(Ia、Ib、Ic)の新しいスルホンアミド誘導体、又はその塩、好ましくはその該当する生理学的に許容される塩、又はその該当する溶媒和物に;その調製のための工程に、ヒト及び/又は動物治療学における医薬品でのその利用に、及びそれを含有する製薬組成物に関するものである。
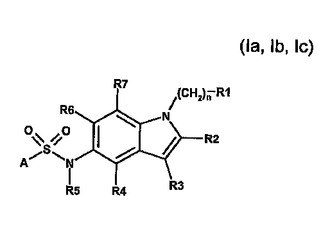 (もっと読む)
(もっと読む)
キナーゼ阻害剤として活性なピリジルピロール誘導体
【課題】 明細書において定義したとおりの式(I)のピリジルピロール誘導体およびこれらの薬学的に許容される塩、並びにこれらを含む薬学的組成物を開示してあり;本発明の化合物は、癌様の、制御傷害されたタンパク質キナーゼ活性と関連した疾患治療の療法において、有用な可能性がある。 (もっと読む)
1,941 - 1,960 / 1,977
[ Back to top ]