
キチンナノファイバー複合材料およびその製造方法
【課題】光学特性、耐熱性、および寸法安定性に優れ、さらには可撓性に優れた複合材料を提供する。
【解決手段】チオール基を有するシルセスキオキサン化合物(a−1)、ならびにエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)を含み、かつ硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)と、平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーからなるキチンナノファイバー不織布(B)と、を含むプリプレグを硬化させてなる、キチンナノファイバー複合材料。
【解決手段】チオール基を有するシルセスキオキサン化合物(a−1)、ならびにエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)を含み、かつ硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)と、平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーからなるキチンナノファイバー不織布(B)と、を含むプリプレグを硬化させてなる、キチンナノファイバー複合材料。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、キチンナノファイバー複合材料およびその製造方法に関する。
【背景技術】
【0002】
一般に、液晶や有機EL素子などのディスプレイ用基板には、ガラス板が広く用いられている。しかし、ガラス板は比重が大きく軽量化が困難であり、さらに割れやすい、曲げられない、所定の厚みが必要などの欠点があることから、近年、ガラス板に代わるプラスチック基板が検討されている。具体的には、ポリカーボネートやポリエチレンテレフタレートなどがディスプレイ基板として使用されている
しかしながら、これら従来のガラス板代替用プラスチック材料は、ガラス板に比べて線膨張率が大きいため、基板上に薄膜トランジスタなどのデバイス層を高温で蒸着させるプロセスの際に、反りや蒸着膜の割れ、半導体の断線などの問題が生じ易く、実使用は困難であった。
【0003】
このような問題に対して、ガラス充填剤またはガラスファイバーによる複合材料(例えば特許文献1、2参照)、セルロースナノファイバー複合材料(例えば特許文献3参照)、部分脱アセチル化処理したキチンナノファイバーを用いた複合材料(例えば特許文献4参照)などが提案されている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2011−52116号公報
【特許文献2】特開2010−155897号公報
【特許文献3】特開2010−180416号公報
【特許文献4】特開2010−180309号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、上記特許文献1および2に記載の技術では、複合材料のマトリックスに用いられる材料とガラス材料とは屈折率の温度依存性が異なるため、使用条件によっては不透明になる可能性が高い。また、ガラス充填剤やガラスファイバーによる複合材料の場合、そのサイズがせいぜいミクロンオーダーであることから、屈曲によってマトリックス材料との界面で剥離が生じやすい。その結果、その部分で光線の散乱が生じ、光学特性、特にヘイズ値を悪化させる。
【0006】
上記特許文献3に記載のナノオーダーであるセルロースナノファイバーとの複合材料においては、上述のような界面剥離の虞は殆どない。しかしながら、セルロース分子の構造上、分子間水素結合が強固であるため、セルロースナノファイバーを生産する工程において、セルロースの分子鎖が切断され、解繊状態が不均一なセルロースナノファイバーとなってしまう。このようなセルロースナノファイバーでは、光学特性、耐熱性、および寸法安定性を十分に満足することはできない。
【0007】
さらに、特許文献4に記載の部分脱アセチル化処理したキチンナノファイバーは、セルロースナノファイバーに比べて繊維径の均一なナノファイバーが得られるが、解繊のために脱アセチル化することは、同時に分子鎖も切断してしまうため、得られる複合材料の耐熱性や寸法安定性が低下してしまう。
【0008】
このように、従来の技術では、光学特性、耐熱性、および寸法安定性に優れ、さらには可撓性に優れた複合材料を得ることは困難であった。
【0009】
そこで、本発明は、光学特性、耐熱性、および寸法安定性に優れ、さらには可撓性に優れた複合材料を提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明者は、上記の問題を解決すべく、鋭意研究を行った。その結果、硬化性樹脂組成物およびキチンナノファイバー不織布を含むプリプレグを硬化させて得られる複合材料が、上記課題を解決することを見出し、本発明を完成するに至った。
【0011】
すなわち、本発明は、チオール基を有するシルセスキオキサン化合物(a−1)、ならびにエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)を含み、かつ硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)と、平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーからなるキチンナノファイバー不織布(B)と、を含むプリプレグを硬化させてなる、キチンナノファイバー複合材料である。
【発明の効果】
【0012】
本発明によれば、光学特性、耐熱性、および寸法安定性に優れ、さらには可撓性に優れたキチンナノファイバー複合材料が提供されうる。
【図面の簡単な説明】
【0013】
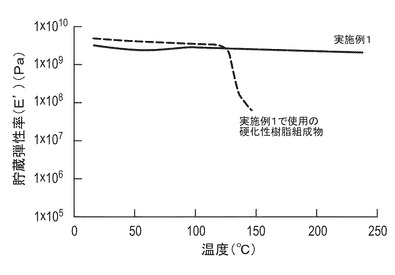
【図1】実施例1で得られたキチンナノファイバー複合材料および実施例1で使用の硬化性樹脂組成物について、温度と動的貯蔵弾性率との関係を示すグラフである。
【発明を実施するための形態】
【0014】
本発明は、チオール基を有するシルセスキオキサン化合物(a−1)、ならびにエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)を含み、かつ硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)と、平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーからなるキチンナノファイバー不織布(B)と、を含むプリプレグを硬化させてなる、キチンナノファイバー複合材料である。
【0015】
以下、本発明のキチンナノファイバー複合材料に含まれる各成分について詳細に説明する。
【0016】
<(A)硬化性樹脂組成物>
(a−1)チオール基を有するシルセスキオキサン化合物
本発明で用いられる硬化性樹脂組成物(以下、単に(A)成分とも称する)に含まれるチオール基を有するシルセスキオキサン化合物(以下、単に成分(a−1)とも称する)は、下記化学式(1)で表されるチオール基含有アルコキシシラン類(a−11)を加水分解および縮合して得られる化合物である。
【0017】
【化1】

【0018】
前記化学式(1)中、R1は少なくとも1つのチオール基を有する炭素数1〜8の炭化水素基、または少なくとも1つのチオール基を有する芳香族炭化水素基を表し、R2は水素原子、炭素数1〜8の炭化水素基、または芳香族炭化水素基を表す。
【0019】
上記チオール基含有アルコキシシラン類(a−11)(以下、単に(a−11)成分とも称する)の具体例としては、例えば、3−メルカプトプロピルトリメトキシシラン、3−メルカプトプロピルトリエトキシシラン、3−メルカプトプロピルトリプロポキシシラン、3−メルカプトプロピルトリブトキシシラン、1,4−ジメルカプト−2−(トリメトキシシリル)ブタン、1,4−ジメルカプト−2−(トリエトキシシリル)ブタン、1,4−ジメルカプト−2−(トリプロポキシシリル)ブタン、1,4−ジメルカプト−2−(トリブトキシシリル)ブタン、2−メルカプトメチル−3−メルカプトプロピルトリメトキシシラン、2−メルカプトメチル−3−メルカプトプロピルトリエトキシシラン、2−メルカプトメチル−3−メルカプトプロピルトリプロポキシシラン、2−メルカプトメチル−3−メルカプトプロピルトリブトキシシラン、1,2−ジメルカプトエチルトリメトキシシラン、1,2−ジメルカプトエチルトリエトキシシラン、1,2−ジメルカプトエチルトリプロポキシシラン、1,2−ジメルカプトエチルトリブトキシシラン等が挙げられる。これらは単独でも、また2種以上組み合わせても用いることができる。これら(a−11)成分のうち、3−メルカプトプロピルトリメトキシシランは、加水分解反応の反応性が高く、かつ入手が容易であるため好ましい。
【0020】
また、上記(a−11)成分に加えて、トリメチルメトキシシラン、トリメチルエトキシシラン、トリエチルメトキシシラン、トリエチルエトキシシラン、トリフェニルメトキシシラン、トリフェニルエトキシシランなどのトリアルキルアルコキシシラン類、ジメチルジメトキシシラン、ジメチルジエトキシシラン、ジエチルジメトキシシラン、ジエチルジエトキシシラン、ジフェニルジメトキシシラン、ジフェニルジエトキシシラン、メチルフェニルジメトキシシラン、メチルフェニルジエトキシシラン、3−メルカプトプロピルメチルジメトキシシランなどのジアルキルジアルコキシシラン類、メチルトリメトキシシラン、メチルトリエトキシシラン、エチルトリメトキシシラン、エチルトリエトキシシラン、フェニルトリメトキシシラン、フェニルトリエトキシシランなどのアルキルトリアルコキシシラン類、テトラメトキシシラン、テトラエトキシシラン、テトラプロポキシシラン、テトラブトキシシランなどのテトラアルコキシシラン類、テトラメトキシチタン、テトラエトキシチタン、テトラプロポキシチタン、テトラブトキシチタンなどのテトラアルコキシチタン類、テトラエトキシジルコニウム、テトラプロポキシジルコニウム、テトラブトキシジルコニウムなどのテトラアルコキシジルコニウム類などの金属アルコキシド類(a−12)(以下、単に(a−12)成分とも称する)を使用することができる。これら(a−12)成分は、単独でも、または2種以上組み合わせても用いることができる。これらのうち、トリアルキルアルコキシシラン類、ジアルキルジアルコキシシラン類、テトラアルコキシシラン類を用いることで、(A)成分の架橋密度を調整することができる。アルキルトリアルコキシシラン類を用いることで、(A)成分中に含まれるチオール基の量を調整することができる。
【0021】
(a−11)成分と(a−12)成分とを併用する場合は、{(a−11)成分に含まれるチオール基のモル数}/{(a−11)成分と(a−12)成分との合計モル数}(1分子あたりに含まれるチオール基の平均個数を示す)が0.2以上であることが好ましい。0.2未満である場合、得られる(a−1)成分中に含まれるチオール基の数が少なくなるため、熱硬化性および紫外線硬化性が低下する傾向がある。また、{(a−11)成分と(a−12)成分とに含まれる各アルコキシ基の合計モル数}/{(a−11)成分と(a−12)成分との合計モル数}(モル比:1分子あたりに含まれるアルコキシ基の平均個数を示す)が2.5以上3.5以下であることが好ましく、2.7以上3.2以下であることがより好ましい。2.5未満の場合、得られる(a−1)成分の架橋密度が低く、複合材料の耐熱性が低下する傾向がある。また、3.5を超える場合、(a−1)成分を製造する際、ゲル化しやすくなる傾向がある。
【0022】
本発明に用いられる(a−1)成分は、(a−11)成分単独やこれに(a−12)成分を併用して、それらを加水分解後、縮合させて得ることができる。加水分解反応によって、(a−11)成分や(a−12)成分に含まれるアルコキシ基が水酸基となり、アルコールが副生する。加水分解反応に必要な水の量は、{加水分解反応に用いる水のモル数}/{(a−11)成分と(a−12)成分とに含まれる各アルコキシ基の合計モル数}(モル比)が0.4以上10以下であればよく、好ましくは0.5以上2以下である。0.4未満の場合、(a−1)成分中に加水分解されずにアルコキシ基が残る虞がある。また、10を超える場合、後に行う縮合反応(脱水反応)の際に除くべき水の量が多くなるため製造時間が長くなり、経済的に不利になる傾向がある。
【0023】
また、(a−12)成分としてテトラアルコキシチタン類やテトラアルコキシジルコニウム類等、特に加水分解性および縮合反応性の高い金属アルコキシド類を併用する場合には、急速に加水分解および縮合反応が進行し、系がゲル化してしまう場合がある。この場合、(a−11)成分の加水分解反応を終了させ、実質的にすべての水が消費された状態にした後、該(a−12)成分を添加することによって、ゲル化を避けることができる。
【0024】
加水分解反応に用いる触媒としては、特に限定されず、従来公知の加水分解触媒を任意に用いることができる。これらのうちギ酸は、触媒活性が高く、また引き続く縮合反応の触媒としても機能するので好ましい。ギ酸の添加量は、(a−11)成分および(a−12)成分の合計100質量部に対して、0.1〜25質量部であることが好ましく、1〜10質量部であることがより好ましい。25質量部よりも多いと、(A)成分の安定性が低下する傾向があり、また後工程でギ酸を除去できるとしても該除去量が多くなる傾向がある。一方、0.1質量部よりも少ないと、実質的に反応が進行しない、または反応時間が長くなるなどの傾向がある。反応温度、反応時間は、(a−11)成分や(a−12)成分の反応性に応じて任意に設定できるが、通常0〜100℃程度、好ましくは20〜60℃、1分〜2時間程度である。該加水分解反応は、溶剤の存在下または不存在下に行うことができる。溶剤の種類は特に限定されず、任意の溶剤を1種類以上選択して用いることができるが、後述の縮合反応に用いる溶剤と同一のものを用いることが好ましい。(a−11)成分や(a−12)成分の反応性が低い場合は、無溶剤で行うことが好ましい。
【0025】
該溶剤の例としては、例えば、トルエン、キシレン、メタノール、エタノール、水、プロピレングリコールモノメチルエーテルアセテート、エチレングリコールジメチルエーテル、トルエン等が挙げられる。
【0026】
上記方法で加水分解反応を行うが、{加水分解されてできた水酸基のモル数}/{(a−11)成分と(a−12)成分とに含まれる各アルコキシ基の合計モル数}(モル比)が0.5以上になるように進行させることが好ましく、0.8以上になるように進行させることがさらに好ましい。
【0027】
縮合反応においては、前記の水酸基間で水が副生し、また水酸基とアルコキシ基との間ではアルコールが副生して、シロキサン結合を生じる。縮合反応には、従来公知の脱水縮合触媒を任意に用いることができる。前記のように、ギ酸は触媒活性が高く、加水分解反応の触媒と共用できるため好ましい。反応温度、反応時間は(a−11)成分や(a−12)成分の反応性に応じてそれぞれ任意に設定できるが、通常は40〜150℃程度、好ましくは60〜100℃、30分〜12時間程度である。
【0028】
上記方法で縮合反応を行うが、{未反応の水酸基および未反応のアルコキシ基の合計モル数}/{(a−11)成分と(a−12)成分とに含まれる各アルコキシ基の合計モル数}(モル比)が0.3以下になるように進行させることが好ましく、0.2以下になるように進行させることがより好ましい。0.3を超える場合、未反応の水酸基およびアルコキシ基が(A)成分の保管中に縮合反応してゲル化したり、硬化後に縮合反応し揮発分が発生してクラックが発生するなど、複合材料の性能を損なう傾向がある。
【0029】
当該縮合反応は、(a−11)成分((a−12)成分を併用する場合は両者)の濃度が2〜80質量%程度になるように溶剤希釈して行うことが好ましく、15〜60質量%であることがより好ましい。縮合反応によって生成する水およびアルコールの沸点より高い沸点を有する溶剤を用いると、反応系中よりこれらを留去することができるため好ましい。該濃度が2質量%未満である場合は、(A)成分に含まれる(a−1)成分が少なくなる傾向がある。80質量%を超える場合は、反応中にゲル化したり、生成する(A)成分の分子量が大きくなり過ぎ、複合材料の保存安定性が悪くなる傾向がある。溶剤としては、任意の溶剤を1種類以上選択して用いることができる。縮合反応によって生成する水およびアルコールより高い沸点を有する溶剤を用いれば、反応系中よりこれらを留去することができるため好ましい。また、後述のエチレン性不飽和結合を有する化合物も溶剤の一部として用いることができる。
【0030】
当該縮合反応の終了後、用いた触媒を除去すると、最終的に得られる複合材料の安定性が向上するため好ましい。除去方法は、用いた触媒に応じて公知各種の方法から適宜に選択できる。例えば、ギ酸を用いた場合は、縮合反応の終了後、該沸点以上に加熱する、減圧するなどの方法により容易に除去でき、この点からもギ酸の使用が好ましい。
【0031】
なお、シルセスキオキサン化合物(a−1)は、ランダム構造、ラダー構造、かご構造、不完全縮合かご構造などの構造を有しうることが知られているが、本発明においては、いずれの構造であっても用いることができる。
【0032】
(a−2)エチレン性不飽和結合を有する化合物またはイソシアネート基を有する化合物
本発明で用いられるエチレン性不飽和結合を有する化合物(以下、単に(a−21)成分とも称する)またはイソシアネート基を有する化合物(以下、単に(a−22)成分とも称する)は、(a−1)成分の硬化剤としての役割を果たす。
【0033】
(a−21)成分中のエチレン性不飽和結合は、エチレン性不飽和結合を有する官能基とチオール基との反応より優先して、エチレン性不飽和結合を有する官能基同士が重合する不都合が起こらないよう、ラジカル重合性が低いものを用いることが好ましい。このような(a−21)成分として、アリル基を1つ以上有する化合物が挙げられる。アリル基を1つ含有する化合物としては、例えば、ケイ皮酸、モノアリルシアヌレート、モノアリルイソシアヌレート、ペンタエリスリトールモノアリルエーテル、トリメチロールプロパンモノアリルエーテル、グリセリンモノアリルエーテル、ビスフェノールAモノアリルエーテル、ビスフェノールFモノアリルエーテル、エチレングリコールモノアリルエーテル、ジエチレングリコールモノアリルエーテル、トリエチレングリコールモノアリルエーテル、プロピレングリコールモノアリルエーテル、ジプロピレングリコールモノアリルエーテル、トリプロピレングリコールモノアリルエーテルなどが挙げられる。アリル基を2つ含有する化合物としては、例えば、ジアリルフタレート、ジアリルイソフタレート、ジアリルシアヌレート、ジアリルイソシアヌレート、ペンタエリスリトールジアリルエーテル、トリメチロールプロパンジアリルエーテル、グリセリンジアリルエーテル、ビスフェノールAジアリルエーテル、ビスフェノールFジアリルエーテル、エチレングリコールジアリルエーテル、ジエチレングリコールジアリルエーテル、トリエチレングリコールジアリルエーテル、プロピレングリコールジアリルエーテル、ジプロピレングリコールジアリルエーテル、トリプロピレングリコールジアリルエーテルなどが挙げられる。アリル基を3つ以上含有する化合物としては、例えば、トリアリルイソシアヌレート、ペンタエリスリトールトリアリルエーテル、ペンタエリスリトールテトラアリルエーテル、トリメチロールプロパントリアリルエーテルなどが挙げられる。これら(a−21)成分は、単独でも、または2種以上組み合わせても用いることができる。これらの中でも、分子中に1つのエチレン性不飽和結合のみを有する化合物では分子間の架橋が起こらないため、複合材料の耐熱性、表面硬度等の物性についての改善効果も不充分となる傾向があることから、分子中に2以上のエチレン性不飽和結合を有する化合物が好ましく、中でもトリアリルイソシアヌレート、ジアリルフタレート、ペンタエリスリトールトリアリルエーテルが特に好ましい。
【0034】
また、(a−21)成分として、上記アリル基を有する化合物よりも高分子量のものを用いることもできる。(a−21)成分として高分子量のものを用いた複合材料は、可撓性が向上する傾向がある。また、一般にラジカル重合性が低くなる傾向があり、このような視点からも好ましく用いることができる。該高分子量物としては、メチルアリルシロキサンとジメチルシロキサンとからなる共重合物、エピクロルヒドリンとアリルグリシジルエーテルとからなる共重合物(ダイソー株式会社製:商品名「エピクロマー(登録商標)」、日本ゼオン株式会社製:商品名「Gechron(登録商標)」など)、アリル基末端ポリイソブチレンポリマー(株式会社カネカ製:商品名「エピオン(登録商標)」)、ウレタンアクリレート(荒川化学工業株式会社製:商品名「ビームセット550B」)などが挙げられる。これらの化合物は、単独でも、または2種以上組み合わせても用いることができる。
【0035】
また、本発明で用いられる(a−22)成分は、特に限定されず、従来公知のイソシアネート基を有する化合物を適宜に用いることができる。該イソシアネート基を有する化合物としては、例えば、芳香族、脂肪族または脂環族の各種公知のジイソシアネート類を使用することができ、より具体的には、例えば、1,5−ナフチレンジイソシアネート、4,4’−ジフェニルメタンジイソシアネート、4,4’−ジフェニルジメチルメタンジイソシアネート、4,4’−ジベンジルイソシアネート、ジアルキルジフェニルメタンジイソシアネート、テトラアルキルジフェニルメタンジイソシアネート、1,3−フェニレンジイソシアネート、1,4−フェニレンジイソシアネート、トリレンジイソシアネート、ブタン−1,4−ジイソシアネート、ヘキサメチレンジイソシアネート、イソプロピレンジイソシアネート、メチレンジイソシアネート、2,2,4−トリメチルヘキサメチレンジイソシアネート、2,4,4−トリメチルヘキサメチレンジイソシアネート、シクロヘキサン−1,4−ジイソシアネート、キシリレンジイソシアネート、水素化キシリレンジイソシアネート、イソホロンジイソシアネート、リジンジイソシアネート、ジシクロヘキシルメタン−4,4’−ジイソシアネート、1,3−ビス(イソシアネートメチル)シクロヘキサン、メチルシクロヘキサンジイソシアネート、m−テトラメチルキシリレンジイソシアネートやダイマー酸のカルボキシル基をイソシアネート基に転化したダイマージイソシアネートなどが挙げられる。これらの化合物は、単独でも、または2種以上組み合わせても用いることができる。該化合物のうち、イソホロンジイソシアネートは、最終的に得られる複合材料が透明性、耐熱性等に優れ、かつ入手が容易であるため特に好ましい。
【0036】
また、(a−22)成分として、前記化合物よりも高分子量のものを用いることができる。高分子量のものを用いてなる複合材料は、可撓性が向上する傾向がある。該高分子量物としては、ポリカーボネートジオール、ポリエステルジオールなどのポリオール類のジイソシアネート変性物、ポリメリックMDI(三井武田ケミカル株式会社製:商品名「コスモネート(登録商標)M」など)、ポリイソシアヌレートタイプのHDI(日本ポリウレタン工業株式会社製:商品名「コロネート(登録商標)HX」など)などが挙げられる。これらの化合物は、単独でも、または2種以上組み合わせても用いることができる。これら化合物のうち、ポリイソシアヌレートタイプのHDIは、最終的に得られる複合材料が透明性、耐熱性等に優れ、かつ入手が容易であるため好ましい。
【0037】
また、高分子量物としてメタクリロイルオキシエチルイソシアネート(昭和電工株式会社製:商品名「カレンズMOI(登録商標)」)、アクリロイルオキシエチルイソシアネート(昭和電工株式会社製:商品名「カレンズAOI(登録商標)」)など、1分子中にエチレン性不飽和結合およびイソシアネート基が共に存在する化合物を用いることもできる。このような化合物を用いた際には、(a−21)成分および(a−22)成分を同時に含有するものとみなすことができ、分子中に含まれるエチレン性不飽和結合の数およびイソシアネート基の数を考慮のうえ使用量を決定する必要がある。
【0038】
また、(a−22)成分を用いる場合には、従来公知のウレタン化触媒を用いることができる。例えば、ジブチルスズジラウレート、オクチル酸スズなどの有機スズ化合物、1,8−ジアザ−ビシクロ[5.4.0]ウンデセン−7、トリエチレンジアミン、ベンジルジメチルアミン、トリエタノールアミン、ジメチルアミノエタノール、トリス(ジメチルアミノメチル)フェノールなどの三級アミン類などをあげることができる。ウレタン化触媒は、(A)成分100質量部に対し、0.01〜5質量部の割合で使用することが好ましい。
【0039】
上記(A)成分における有効(a−1)成分、(a−21)成分および(a−22)成分の濃度は、用途に応じて適宜に決定でき、必要に応じて溶剤を配合することができる。溶剤としては、当該成分と非反応性であればよく、各種従来公知のものを適宜選択して用いることができる。(A)成分を1mm以上の厚膜に硬化させる場合は、(a−1)成分、(a−21)成分および(a−22)成分の合計濃度を(A)成分中90質量%以上にすることが好ましく、95質量%以上にすることがより好ましい。該合計濃度は、(a−1)成分、(a−21)成分および(a−22)成分の濃度と(A)成分の仕込み時に加えた溶剤の量により計算で求めることができ、また(A)成分に含まれる溶剤の沸点以上で2時間程度加熱し、加熱前後の重量変化により求めることもできる。(A)成分を1mm以上の厚膜に硬化させる場合は、90質量%未満の場合、硬化、成型時に発泡したり、複合材料中に溶剤が残存したりして、複合材料の物性が低下する傾向がある。なお、(a−1)成分を合成する際に溶剤を使用しているため、反応終了後、不揮発分含有量が90質量%以上となるよう溶剤を揮発させておけばよい。また、(A)成分を調製した後、用いた溶剤を揮発させて、有効な(a−1)成分、(a−21)成分および(a−22)成分の合計濃度を高めることもできる。
【0040】
上記(A)成分は、(a−1)成分、ならびに(a−21)成分および(a−22)成分の少なくとも一方を含有するものである。(A)成分中に含有される各成分は、[{(a−21)成分中に含まれるエチレン性不飽和結合の数}/{(a−1)成分中のチオール基の数}]が0.1〜0.8、[{(a−22)成分中のイソシアネート基の数}/{(a−1)成分中のチオール基の数}]が0.1〜0.8、[{(a−21)成分中に含まれるエチレン性不飽和結合の数+(a−22)成分中に含まれるイソシアネート基の数}/{(A)成分中のチオール基の数}]が0.9〜1.1なる割合で含有することが好ましい。
【0041】
[{(a−21)成分中に含まれるエチレン性不飽和結合の数+(a−22)成分中に含まれるイソシアネート基の数)}/{(a−1)成分中のチオール基の数}]が0.9未満の場合には、チオール基が残存し、その分解によって悪臭を発生させる場合がある。1.1を超える場合には、硬化後にエチレン性不飽和結合やイソシアネート基が残存し、耐候性が低下する傾向がある。
【0042】
[{(a−21)成分中に含まれるエチレン性不飽和結合の数}/{(a−1)成分中のチオール基の数}]が0.8を超えると、紫外線硬化を第一段階とする場合には硬化が進行しすぎ、熱硬化を第一段階とする場合には半硬化物中に未反応の(a−21)成分が多くなりすぎるため、いずれも成型加工性が失われる場合がある。また、0.1未満の場合は、成型加工性が失われる場合がある。
【0043】
{(a−22)成分中のイソシアネート基の数}/{(a−1)成分中のチオール基の数)}が0.8を超えると、紫外線硬化を第一段階とする場合には半硬化物中に未反応の(a−22)成分が多くなりすぎるため、熱硬化を第一段階とする場合には硬化が進行しすぎ、いずれも成形加工性が失われる場合がある。また、0.1未満の場合は、成形加工性が失われる場合がある。
【0044】
また、(A)成分には、用途に応じ、上記(a−11)成分および/またはその加水分解物(以下、併せて(E)成分とも称する)を配合できる。(E)成分は、(a−1)成分の合成に際して用いた(a−11)成分をそのままで用いるか、その加水分解物を用いるか、これらを組み合わせて使用できる。(E)成分の配合量は、(A)成分100質量部に対して、0.1〜20質量部程度であることが好ましい。0.1質量部未満の場合は、複合材料の無機基材に対する密着性向上効果が不充分となる傾向がある。また、20質量部を超える場合、(E)成分が加水分解、縮合反応する際の揮発分が多くなるため、(A)成分が硬化時に発泡したり、反りやクラックが発生したり、得られる複合材料が脆くなったりする傾向がある。このような(E)成分としては、3−メルカプトプロピルトリメトキシシランが、密着性向上効果の点で特に好ましい。
【0045】
さらに、(A)成分には、用途に応じ、上記(a−12)成分である金属アルコキシド類および/またはその加水分解物(以下、併せて(F)成分とも称する)を配合できる。(F)成分は、(a−1)成分の合成に際して用いた金属アルコキシド類をそのままで用いるか、その加水分解物を用いるか、これらを組み合わせて使用できる。(F)成分を含有する(A)成分を用いることで、得られる複合材料の屈折率を調整することができる。(F)成分の配合量は、(A)成分100質量部に対して、0.1〜20質量部程度であることが好ましい。0.1質量部に満たない場合には、屈折率向上効果が不充分となる傾向がある。また、20質量部を超える場合は、(F)成分が加水分解、縮合反応する際の揮発分が多くなるため、(A)成分が硬化時に発泡したり、反りやクラックが発生したり、得られる複合材料が脆くなったりする傾向がある。
【0046】
(A)成分は、熱硬化用触媒および/または光硬化用触媒を含むことが好ましい。前記熱硬化用触媒の例としては、従来公知のウレタン化触媒を用いることができる。例えば、ジブチル錫ジラウリレート、オクチル酸錫などの有機錫化合物、1,8−ジアザ−ビシクロ[5.4.0]ウンデセン−7、トリエチルジアミン、ベンジルメチルアミン、トリエタノールアミン、ジメチルアミノエタノール、トリス(ジメチルアミノメチル)フェノールなどの三級アミン類などをあげることができる。また、前記光硬化用触媒の例としては、従来公知の光カチオン開始剤、光ラジカル開始剤などを任意に選択できる。光カチオン開始剤としては、紫外線の照射により酸を発生する化合物であるスルホニウム塩、ヨードニウム塩、メタロセン化合物、ベンゾイントシレート等があげられ、それらの市販品としては、例えば、サイラキュア(登録商標)UVI−6970、同UVI−6974、同UVI−6990(いずれも米国ユニオンカーバイド社製商品名)、イルガキュア(登録商標)264(チバスペシャルティケミカルズ社製)、CIT−1682(日本曹達株式会社などが挙げられる。光ラジカル開始剤としては、ダロキュア(登録商標)1173、イルガキュア(登録商標)651、イルガキュア(登録商標)184、イルガキュア(登録商標)907(いずれもチバスペシャルティケミカルズ社製商品名)、ベンゾフェノン等が挙げられる。なお、得られる硬化物の耐候性低下が懸念される場合、特に高い耐候性、透明性が求められる光学部材などに用いられる場合には、光反応開始剤や光増感剤を使用しないほうがよい。
【0047】
また、硬化性樹脂組成物(A)の安定性をより向上させるため、エン−チオール反応を抑制する化合物を配合できる。このような化合物としては、トリフェニルホスフィン、亜リン酸トリフェニル等のリン系化合物;p−メトキシフェノール、ハイドロキノン、ピロガロ−ル、ナフチルアミン、tert−ブチルカテコール、塩化第一銅、2,6−ジ−tert−ブチル−p−クレゾール、2,2’−メチレンビス(4−エチル−6−tert−ブチルフェノール)、2,2’−メチレンビス(4−メチル−6−tert−ブチルフェノール)、N−ニトロソフェニルヒドロキシルアミンアルミニウム塩、ジフェニルニトロソアミン等のラジカル重合禁止剤;ベンジルジメチルアミン、2−(ジメチルアミノメチル)フェノール、2,4,6−トリス(ジアミノメチル)フェノール、ジアザビシクロウンデセン等の3級アミン類;2−メチルイミダゾール、2−エチル−4−メチルイミダゾール、2−エチルへキシルイミダゾール、2−ウンデシルイミダゾール、1−シアノエチル−2−メチルイミダゾール等のイミダゾール類があげられる。
【0048】
(A)成分には、本発明の効果を損なわない範囲で、各種用途での必要性に応じて、可塑剤、耐候剤、酸化防止剤、熱安定剤、滑剤、帯電防止剤、増白剤、着色剤、導電剤、離型剤、表面処理剤、粘度調節剤、フィラー等を配合してもよい。
【0049】
<硬化性樹脂組成物の製造方法>
本発明に用いられる硬化性樹脂組成物の製造方法は、特に制限されないが、上記の(a−1)成分、(a−2)成分、ならびに必要に応じて熱硬化触媒、光硬化触媒、および添加剤などを、溶媒中で一括に混合するか、各成分を順次混合するか、または任意の複数の成分を混合した後に残りの成分を混合するなどして、均一な混合物となるように攪拌する方法が挙げられる。
【0050】
調製に用いる溶媒としては、例えば、エチレングリコールジメチルエーテル、トルエン等が挙げられる。
【0051】
(A)成分の調製は、例えば、スターラーなどで均一になるまで、10〜50℃の温度範囲で、5〜60分攪拌することにより行うことができる。
【0052】
このようにして得られる硬化性樹脂組成物(A)の硬化後の屈折率は、1.53〜1.57である。屈折率が1.53未満の場合には、キチンナノファイバー不織布との屈折率の違いが大きく、最終的に得られる複合材料のヘイズが低下する。一方、1.57を超える場合には、複屈折が生じ、最終的に得られる複合材料のヘイズが低下する。該屈折率は、好ましくは1.532〜1.565である。
【0053】
該屈折率は、硬化性樹脂組成物(A)に含まれる各成分の配合比を調整することにより、所望の値とすることができる。また、該屈折率は、実施例に記載の方法により測定することができる。
【0054】
<キチンナノファイバー不織布>
本発明で用いられるキチンナノファイバー不織布は、平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーを絡合して得られるものである。
【0055】
前記平均繊維径が45nm未満であると、キチンナノファイバー不織布に硬化性樹脂組成物を空隙なく含浸させることが困難となり、空隙による光散乱が生じ、その結果複合材料のヘイズが悪化する。一方、65μmを超えると、キチンナノファイバーによる補強効果が薄れ、複合材料の耐熱性および寸法安定性が悪化する。繊維径分布が1.3を超えると、繊維径の大きな部分での光散乱が生じ、その結果複合材料のヘイズが悪化する。該平均繊維径は好ましくは41〜48nmであり、該繊維径分布は好ましくは1.1〜1.28である。なお、繊維径分布は、定義上、1未満の値にはならない。該平均繊維径および該繊維径分布は、実施例に記載の方法により測定することができる。
【0056】
上記のような平均繊維径および繊維径分布を有するキチンナノファイバーを得る方法は特に制限されないが、例えば、市販の精製キチンを酸性液体に浸漬させた後、解繊処理を行う方法が挙げられる。
【0057】
前記酸性液体としては、所望の範囲のpHが得られる限度で任意の酸を用いることができる。すなわち、酸は、有機酸であってもよく、無機酸であってもよく、特に制限されない。また、酸性液体の溶媒にも特に限定はなく、水以外のものを用いてもよい。
【0058】
前記有機酸としては、例えば、ギ酸、酢酸、クエン酸、リンゴ酸、シュウ酸、サリチル酸、アスコルビン酸、酒石酸、グルコン酸、乳酸、フマル酸、コハク酸、コハク酸ナトリウム、フィチン酸、アジピン酸、プロピオン酸、グリオキシル酸、ピルビン酸、アセト酢酸、レブリン酸、ヘプタン酸、カプリル酸、カプリン酸、ラウリル酸、グリコール酸、グリセリン酸、アクリル酸、安息香酸、パラニトロ安息香酸、パラトルエンスルホン酸、ピクリン酸、マレイン酸などが挙げられる。前記無機酸としては、例えば、リン酸、塩酸、硫酸、硝酸、ピロリン酸二水素二ナトリウムなどが挙げられる。
【0059】
前記酸性液体のpHは5以下であることが好ましい。かような範囲であれば、上記範囲の平均繊維径および繊維径分布を有するキチンナノファイバーを効率よく得ることができる。
【0060】
前記解繊処理は、例えば、家庭用ミキサー(プロペラミキサー、カッターミキサー)、超音波ホモジナイザー、高圧ホモジナイザー、二軸混練機、石臼式粉砕機などの解繊・粉砕装置を用いて行うことができる。
【0061】
上記方法によりキチンナノファイバーを得た後、絡合処理を行うことにより、キチンナノファイバー不織布を得ることができる。絡合処理の方法も特に制限されず、乾式法や湿式法など、いずれの方法で行ってもよい。
【0062】
得られたキチンナノファイバー不織布に、上記硬化性樹脂組成物を含浸させプリプレグを作製し、該プリプレグを硬化させることにより、本発明の複合材料が得られる。プリプレグを得る方法、およびプリプレグを硬化させる方法については、後述する。
【0063】
<キチンナノファイバー複合材料の製造方法>
本発明のキチンナノファイバー複合材料の製造方法は特に制限されないが、下記の工程1〜4を含むことが好ましい。
【0064】
(工程1)チオール基を有するシルセスキオキサン化合物(a−1)とエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)とを混合し、屈折率が1.53〜1.57である硬化性樹脂組成物(A)を得る工程。
【0065】
(工程2)平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーを絡合させてキチンナノファイバー不織布(B)を得る工程。
【0066】
(工程3)前記硬化性樹脂組成物(A)を前記キチンナノファイバー不織布(B)に含浸させてプリプレグを得る工程。
【0067】
(工程4)前記プリプレグを硬化させる工程。
【0068】
これらの工程の中でも、工程1および工程2は、上記で説明した通りであるため、ここでは説明を省略する。以下では、工程3および工程4について説明する。
【0069】
(工程3)
本工程では、硬化性樹脂組成物(A)をキチンナノファイバー不織布(B)に含浸させてプリプレグを作製する。
【0070】
硬化性樹脂組成物(A)をキチンナノファイバー不織布(B)に含浸させる方法は特に制限されないが、例えば、浸漬などによりキチンナノファイバー不織布(B)内部に均一に含浸させる方法、キチンナノファイバー不織布(B)の表面と裏面とに硬化性樹脂組成物(A)を塗布する方法などが挙げられる。
【0071】
キチンナノファイバー不織布(B)に対する硬化性樹脂組成物(A)の使用割合は、特に制限されないが、通常はキチンナノファイバー不織布(B)100質量部あたり20〜500質量部程度である。
【0072】
(工程4)
本工程では、工程3で得られたプリプレグを硬化させ、キチンナノファイバー複合材料を得る。
【0073】
硬化の前に、プリプレグを加熱して溶剤乾燥を行うことが好ましい。加熱乾燥の条件は特に制限されないが、通常70〜100℃の温度で、2〜15分間行われる。
【0074】
加熱して溶剤を乾燥させた後、熱硬化および紫外線硬化の少なくとも一方を行う。
【0075】
前記熱硬化は、90〜130℃の温度で、5〜30分間行うことが好ましい。また、紫外線硬化の際の積算光量は250〜2000mJ/cm2であることが好ましい。
【0076】
熱硬化および紫外線硬化の両方を行う場合は、一段階目で熱硬化を行った後二段階目で紫外線硬化を行ってもよいし、一段階目で紫外線硬化を行った後二段階目で熱硬化を行ってもよい。
【0077】
上記硬化が終了した後は、必要に応じてアニール処理を行う。アニール処理の条件も特に制限されず、通常100〜180℃の温度範囲で、10〜60分間加熱処理することにより行われる。
【0078】
こうして得られる本発明のキチンナノファイバー複合材料は、例えば、液晶パネルや有機ELディスプレイなどのディスプレイ用基板、導光板、偏光板、PDPパネル、OHPフィルム、光ファイバー、カラーフィルター、光ディスク基板、レンズ、液晶セル用プラスチック基板などに好適に用いられる。
【実施例】
【0079】
本発明を、以下の実施例および比較例を用いてさらに詳細に説明する。ただし、本発明の技術的範囲が以下の実施例のみに制限されるわけではない。なお、屈折率、平均繊維径、および繊維径分布の測定は、以下の方法で行った。
【0080】
・屈折率:株式会社アタゴ製、アッベ屈折率計 DR−M2を用いて、波長589nmの屈折率を測定した。
【0081】
・平均繊維径および繊維径分布:ナノファイバー不織布を走査型電子顕微鏡で観察、写真撮影を行い、得られた写真に直線を数本引き、この線を横切る繊維の直径を1枚の写真から200本読み取る。これを、それぞれのナノファイバー不織布について5回繰り返し、合計で1,000本の繊維径のデータから平均繊維径および繊維径分布を求める。それぞれの算出方法は、次の式に従った。
【0082】
【数1】
【0083】
(a−1)チオール基を有するシルセスキオキサン化合物
(a−1−1)の合成
攪拌機、冷却管、分水器、温度計、窒素吹き込み口を備えた反応装置に、3−メルカプトプロピルトリメトキシシラン(信越化学工業株式会社製、商品名「KBM−803」)3400質量部、イオン交換水936質量部、95%ギ酸68質量部を仕込み、室温(25℃)で30分間加水分解反応させた。反応中、発熱によって最大35℃温度上昇した。反応後、トルエン5670質量部を仕込み、加熱した。71℃まで昇温したところで、加水分解によって発生したメタノールと、トルエンの一部が留去され始めた。2時間かけて75℃まで昇温し、縮合反応させて水を留去した。さらに1時間、75℃で反応させた後、70℃、200hPaで減圧して、残存するトルエンの一部、メタノール、水、およびギ酸を留去した。さらに70℃、7hPaで減圧してトルエンを留去することで、チオール基を含有するシルセスキオキサン化合物(a−1−1)を2330質量部得た。濃度は99.0%であった。また、(a−1−1)のチオール基の濃度は、7.41ミリモル/gであった。
【0084】
(a−1−2)の合成
(a−1−1)の合成で用いた装置と同様の装置に、3−メルカプトプロピルトリメトキシシラン15.0質量部、フェニルトリメトキシシラン(東京化成工業株式会社製)5.05質量部、イオン交換水5.51質量部、95%ギ酸1.00質部を仕込み、室温(25℃)で30分間加水分解反応させた。反応中、発熱によって最大20℃温度上昇した。反応後、プロピレングリコールモノメチルエーテルアセテート19.52質量部を仕込み、加熱した。82℃まで昇温したところで、加水分解によって発生したメタノールが留去され始めた。30分かけて105℃まで昇温し、縮合反応させて水を留去した。さらに1時間30分、105℃で反応させた後、70℃、20kPa(150mmHg)で減圧して、残存するメタノール、水、ギ酸を留去することで、チオール基を含有するシルセスキオキサン化合物(a−1−2)を25.13質量部得た。濃度は51.8%であった。また、縮合物(a−1−2)のチオール当量は、329g/eqであった。
【0085】
(a−1−3)の合成
(a−1−1)の合成で用いた装置と同様の装置に、3−メルカプトプロピルトリメトキシシラン180質量部、イオン交換水49.55質量部、95%ギ酸9.00質量部を仕込み、室温(25℃)で30分間加水分解反応させた。反応中、発熱によって最大22℃温度上昇した。反応後、トルエン272.23質量部を仕込み、加熱した。72℃まで昇温したところで、加水分解によって発生したメタノールとトルエンの一部が留去され始めた。20分かけて75℃まで昇温し、縮合反応させて水を留去した。さらに1時間、75℃で反応させた後、70℃、20kPa(150mmHg)で減圧して、残存するメタノール、水、ギ酸を留去した。さらに70℃、0.67kPa(5mmHg)で減圧して、トルエンを留去することで、チオール基を含有するシルセスキオキサン化合物(a−1−3)を124.49質量部得た。濃度は93.7%であった。また、シルセスキオキサン化合物(a−1−3)のチオール当量は、136g/eqであった。
【0086】
(a−1−4)の合成
(a−1−1)の合成で用いた装置と同様の装置に、3−メルカプトプロピルトリメトキシシラン(東レ・ダウコーニング株式会社製、商品名「SH−6062」)190質量部、イオン交換水52.3質量部、95%ギ酸9.5質量部を仕込み、室温(25℃)で30分間加水分解反応させた。反応中、発熱によって最大22℃温度上昇した。反応後、プロピレングリコールモノメチルエーテルアセテート(日本乳化剤株式会社製、商品名「MFG−AC」)287.36質量部を仕込み、加熱した。82℃まで昇温したところで、加水分解によって発生したメタノールが留去され始めた。30分かけて105℃まで昇温し、縮合反応によって発生した水を留去した。さらに1時間30分、105℃で反応させた後、70℃、20kPa(150mmHg)で減圧して、残存するメタノール、水、ギ酸、およびプロピレングリコールモノメチルエーテルアセテートの一部を留去することで、チオール基を含有するシルセスキオキサン化合物(a−1−4)を385.2g得た。濃度は32.0%であった。また、(a−1−4)のチオール当量は、398g/eqであった。
【0087】
(A)硬化性樹脂組成物の製造
(A−1)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−1)31.7質量部に対して、硬化剤である(a−2)成分としてトリアリルイソシアヌレート(以下TAICとも称する、日本化成株式会社製、商品名「TAIC」、エチレン性不飽和結合の濃度は12.0ミリモル/g)5.4質量部、および多官能イソシアネート(日本ポリウレタン株式会社製、商品名「コロネート(登録商標)HX」、イソシアネート基の濃度は5.00ミリモル/g)33.0質量部、熱硬化用触媒としてジブチルスズジラウレート(以下U−100とも称する、日東化成株式会社製、商品名「ネオスタンU−100」)0.15質量部、光硬化用触媒として1−ヒドロキシシクロヘキシルフェニルケトン(以下Irg184とも称する、チバスペシャルティケミカルズ株式会社製、商品名「イルガキュア(登録商標)184」)0.15質量部、および希釈溶剤としてエチレングリコールジメチルエーテル29.6質量部を配合した。25℃で15分間攪拌混合し、硬化性樹脂組成物(A−1)を得た。
【0088】
(A−2)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−2)23.9質量部に対し、硬化剤である(a−2)成分としてTAIC6.0質量部、亜リン酸トリフェニル(東京化成工業株式会社製)0.4質量部、および希釈溶剤としてエチレングリコールジメチルエーテル69.7質量部を配合し、25℃で15分間攪拌混合し、硬化性樹脂組成物(A−2)を得た。
【0089】
(A−3)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−3)18.6質量部に対し、硬化剤である(a−2)成分としてTAIC11.4質量部、亜リン酸トリフェニル(東京化成工業株式会社製)0.4質量部、および希釈溶剤としてエチレングリコールジメチルエーテル69.6質量部を配合し、25℃で15分間攪拌混合し、硬化性樹脂組成物(A−3)を得た。
【0090】
(A−4)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−1)20質量部に対し、硬化剤である(a−2)成分として多官能ウレタンアクリレート(以下550Bとも称する、荒川化学工業株式会社製、商品名「ビームセット550B」、炭素−炭素二重結合の濃度は2.25ミリモル/g)38.1質量部、多官能イソシアネートとしてコロネート(登録商標)HXを11.8質量部、熱硬化用触媒としてU−100を0.15質量部、光硬化用触媒としてIrg184を0.15質量部、および希釈溶剤としてエチレングリコールジメチルエーテル29.8質量部を配合し、25℃で15分間攪拌混合し、硬化性樹脂組成物(A−4)を得た。
【0091】
(A−5)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−4)23.8質量部に対し、硬化剤である(a−2)成分として550Bを4.9質量部、亜リン酸トリフェニル(東京化成工業株式会社製)0.4質量部、屈折率調整剤としてテトラブトキシチタン1.4質量部、および希釈溶剤としてエチレングリコールジメチルエーテル69.5質量部を配合し、25℃で15分間攪拌混合し、硬化性樹脂組成物(A−5)を得た。
【0092】
(B)キチンナノファイバー不織布
(B−1)の製造
蒸留水100質量部に、精製キチン(ナカライテスク株式会社製)1.5質量部、氷酢酸(キシダ化学株式会社製)0.5質量部を添加し、pHを3に調整した。これを、石臼式粉砕機(増幸産業株式会社製、スーパーマスコロイダー MKCA6−2)で解繊処理を1回行うことによってキチンナノファイバーのペースト状物を得た。このペースト状物100質量部に蒸留水500質量部を加えて希釈液とし、この希釈液を親水性PTFEメンブレンフィルター(日本ミリポア株式会社製、オムニポア 平均空孔径1μm)を用いて吸引濾過することによって、親水性PTFEメンブレンフィルター上にキチンナノファイバー不織布(B−1)を得た。得られたキチンナノファイバー不織布(B−1)の厚みは55μm、空孔率は52%、平均繊維径は48nm、繊維径分布は1.13であった。
【0093】
(B−2)の製造
氷酢酸の量を0.4質量部としpHを4に調整すること以外は、上記(B−1)と同様にして、キチンナノファイバー不織布(B−2)を得た。得られたキチンナノファイバー不織布(B−2)の厚みは52μm、空孔率は49%、平均繊維径は61nm、繊維径分布は1.28であった。
【0094】
(B−3)の製造
氷酢酸の量を0.2質量部としpHを6に調整すること以外は、上記(B−1)と同様にして、キチンナノファイバー不織布(B−3)を得た。得られたキチンナノファイバー不織布(B−3)の厚みは55μm、空孔率は51%、平均繊維径は62nm、繊維径分布は1.35であった。
【0095】
(B−4)の製造
上記(B−1)に記載の1度解繊処理をして得られたキチンナノファイバーペースト状物を、石臼式粉砕機を用いてさらに繰り返し2回解繊処理を加えること以外は、上記(B−1)と同様にして、キチンナノファイバー不織布(B−4)を得た。得られたキチンナノファイバー不織布(B−4)の厚みは48μm、空孔率は47%、平均繊維径は41nm、繊維径分布は1.10であった。
【0096】
(D−1)セルロースナノファイバー不織布の製造
濾紙(アドバンテック社製のFILTER PAPER)をハサミで3mm角に切断したもの2gを200mlのビーカに入れた。そこに、N,N−ジメチルアセトアミド 50mLとイオン液体である塩化1−ブチル−3−メチルイミダゾリウム60gを加え、80℃で磁性攪拌子を用いて60分攪拌した後、6モルの硫酸水溶液9質量部を加え、90℃で60分加水分解反応を行った。その後、反応液を室温(25℃)まで冷却してから100質量部のN,N−ジメチルアセトアミドを投入し、攪拌して分散液とした。その後、遠心分離機を用いて、分散液を蒸留水で置換、洗浄することによって、1−ブチル−3−メチルイミダゾリウム、N−ジメチルアセトアミド、硫酸、および水溶性糖分などを除去したセルロースナノファイバー分散液(固形分濃度0.2質量%)を得た。
【0097】
この分散液を親水性PTFEメンブレンフィルター(日本ミリポア株式会社製、オムニポア 平均空孔径1μm)を用いて吸引濾過することによって、親水性PTFEメンブレンフィルター上にセルロースナノファイバー不織布(D−1)を得た。得られたセルロースナノファイバー不織布(D−1)の厚みは56μm、空孔率は58%、平均繊維径は55nm、繊維径分布は1.45であった。
【0098】
(実施例1)
硬化性樹脂組成物(A−1)をキチンナノファイバー不織布(B−1)に含浸させプリプレグを得た後、PETフィルム上に静置し、ホットプレートを用いて80℃で5分、および110℃で10分の条件で、溶剤乾燥および熱硬化を行った。さらに、紫外線照射装置を用いて254nmの検出器で積算光量が500mJ/cm2となるよう紫外線を照射し紫外線硬化を行った後、120℃のオーブン中で15分間アニールを行い、膜厚75μmの透明なキチンナノファイバー複合材料を得た。なお、(A−1)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.554であった。
【0099】
(実施例2)
硬化性樹脂組成物(A−2)をキチンナノファイバー不織布(B−1)に含浸させプリプレグを得た後、PETフィルム上に静置し、ホットプレートを用いて80℃で5分間溶剤乾燥を行った。次に、紫外線照射装置を用いて254nmの検出器で積算光量が500mJ/cm2となるよう紫外線を照射し紫外線硬化を行った後、120℃のオーブン中で15分間アニールを行い、膜厚73μmの透明なキチンナノファイバー複合材料を得た。なお、(A−2)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.532であった。
【0100】
(実施例3)
硬化性樹脂組成物として(A−3)を使用すること以外は、実施例2と同様の方法により、膜厚78μmの透明なキチンナノファイバー複合材料を得た。なお、(A−3)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.565であった。
【0101】
(比較例1)
硬化性樹脂組成物として(A−4)を使用すること以外は、実施例1と同様の方法により、膜厚76μmの透明なキチンナノファイバー複合材料を得た。なお、(A−4)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.520であった。
【0102】
(比較例2)
硬化性樹脂組成物として(A−5)を使用すること以外は、実施例2と同様の方法により、膜厚72μmの透明なキチンナノファイバー複合材料を得た。なお、(A−5)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.594であった。
【0103】
(実施例4)
キチンナノファイバー不織布として(B−2)を使用すること以外は、実施例1と同様の方法により、膜厚74μmの透明なキチンナノファイバー複合材料を得た。
【0104】
(比較例3)
キチンナノファイバー不織布として(B−3)を使用すること以外は、実施例1と同様の方法により、膜厚78μmの透明なキチンナノファイバー複合材料を得た。
【0105】
(比較例4)
キチンナノファイバー不織布として(B−4)を使用すること以外は、実施例1と同様な操作を行い膜厚75μmの透明なキチンナノファイバー複合材料を得た。
【0106】
(比較例5)
キチンナノファイバー不織布の代わりにセルロースナノファイバー不織布(D−1)を用いること以外は、実施例1と同様の方法により、膜厚72μmの透明なセルロースナノファイバー複合材料を得た。
【0107】
<評価>
・寸法安定性:株式会社リガク製、TMA8310を用いて、窒素雰囲気下、1分間に5℃の割合で室温(25℃)から250℃まで昇温させ、荷重45mN、引っ張りモードで寸法変化を測定し、27℃から200℃の温度範囲における線膨張係数を算出した。
【0108】
・全光線透過率:株式会社島津製作所製、分光光度計 UV−2200(積分球付き)を用い、380nmから780nmの波長範囲における全光線透過率(Tt)を測定した。
【0109】
・ヘイズ:株式会社島津製作所製、分光光度計 UV−2200(積分球付き)を用い、380nmから780nmの波長範囲における拡散透過率(Td)を測定し、次の式に従いヘイズを算出した。
【0110】
【数2】
【0111】
各実施例および比較例の上記評価方法の結果を下記表1に示す。
【0112】
【表1】
【0113】
上記表1から明らかなように、本発明の複合材料(実施例1〜4)は、本発明の範囲外である複合材料(比較例1〜5)と比べて、寸法安定性およびヘイズに優れる。また、本発明のキチンナノファイバー複合材料は、ガラス材料を含む複合材料に比べて可撓性に優れる。
【0114】
また、実施例1で得られたキチンナノファイバー複合材料および実施例1で用いられた硬化性樹脂組成物の耐熱性を評価した。具体的には、セイコーインスツルメント株式会社製、粘弾性測定器DMS6100を用い、1分間に5℃の割合で室温(25℃)から250℃まで昇温させ、測定周波数1Hzで動的貯蔵弾性率を測定した。その結果を図1に示す。図1から明らかなように、実施例1のキチンナノファイバー複合材料は、硬化性樹脂組成物に比べて高温での弾性率の低下が少なく、耐熱性に優れていることが認められる。
【技術分野】
【0001】
本発明は、キチンナノファイバー複合材料およびその製造方法に関する。
【背景技術】
【0002】
一般に、液晶や有機EL素子などのディスプレイ用基板には、ガラス板が広く用いられている。しかし、ガラス板は比重が大きく軽量化が困難であり、さらに割れやすい、曲げられない、所定の厚みが必要などの欠点があることから、近年、ガラス板に代わるプラスチック基板が検討されている。具体的には、ポリカーボネートやポリエチレンテレフタレートなどがディスプレイ基板として使用されている
しかしながら、これら従来のガラス板代替用プラスチック材料は、ガラス板に比べて線膨張率が大きいため、基板上に薄膜トランジスタなどのデバイス層を高温で蒸着させるプロセスの際に、反りや蒸着膜の割れ、半導体の断線などの問題が生じ易く、実使用は困難であった。
【0003】
このような問題に対して、ガラス充填剤またはガラスファイバーによる複合材料(例えば特許文献1、2参照)、セルロースナノファイバー複合材料(例えば特許文献3参照)、部分脱アセチル化処理したキチンナノファイバーを用いた複合材料(例えば特許文献4参照)などが提案されている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2011−52116号公報
【特許文献2】特開2010−155897号公報
【特許文献3】特開2010−180416号公報
【特許文献4】特開2010−180309号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、上記特許文献1および2に記載の技術では、複合材料のマトリックスに用いられる材料とガラス材料とは屈折率の温度依存性が異なるため、使用条件によっては不透明になる可能性が高い。また、ガラス充填剤やガラスファイバーによる複合材料の場合、そのサイズがせいぜいミクロンオーダーであることから、屈曲によってマトリックス材料との界面で剥離が生じやすい。その結果、その部分で光線の散乱が生じ、光学特性、特にヘイズ値を悪化させる。
【0006】
上記特許文献3に記載のナノオーダーであるセルロースナノファイバーとの複合材料においては、上述のような界面剥離の虞は殆どない。しかしながら、セルロース分子の構造上、分子間水素結合が強固であるため、セルロースナノファイバーを生産する工程において、セルロースの分子鎖が切断され、解繊状態が不均一なセルロースナノファイバーとなってしまう。このようなセルロースナノファイバーでは、光学特性、耐熱性、および寸法安定性を十分に満足することはできない。
【0007】
さらに、特許文献4に記載の部分脱アセチル化処理したキチンナノファイバーは、セルロースナノファイバーに比べて繊維径の均一なナノファイバーが得られるが、解繊のために脱アセチル化することは、同時に分子鎖も切断してしまうため、得られる複合材料の耐熱性や寸法安定性が低下してしまう。
【0008】
このように、従来の技術では、光学特性、耐熱性、および寸法安定性に優れ、さらには可撓性に優れた複合材料を得ることは困難であった。
【0009】
そこで、本発明は、光学特性、耐熱性、および寸法安定性に優れ、さらには可撓性に優れた複合材料を提供することを目的とする。
【課題を解決するための手段】
【0010】
本発明者は、上記の問題を解決すべく、鋭意研究を行った。その結果、硬化性樹脂組成物およびキチンナノファイバー不織布を含むプリプレグを硬化させて得られる複合材料が、上記課題を解決することを見出し、本発明を完成するに至った。
【0011】
すなわち、本発明は、チオール基を有するシルセスキオキサン化合物(a−1)、ならびにエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)を含み、かつ硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)と、平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーからなるキチンナノファイバー不織布(B)と、を含むプリプレグを硬化させてなる、キチンナノファイバー複合材料である。
【発明の効果】
【0012】
本発明によれば、光学特性、耐熱性、および寸法安定性に優れ、さらには可撓性に優れたキチンナノファイバー複合材料が提供されうる。
【図面の簡単な説明】
【0013】
【図1】実施例1で得られたキチンナノファイバー複合材料および実施例1で使用の硬化性樹脂組成物について、温度と動的貯蔵弾性率との関係を示すグラフである。
【発明を実施するための形態】
【0014】
本発明は、チオール基を有するシルセスキオキサン化合物(a−1)、ならびにエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)を含み、かつ硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)と、平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーからなるキチンナノファイバー不織布(B)と、を含むプリプレグを硬化させてなる、キチンナノファイバー複合材料である。
【0015】
以下、本発明のキチンナノファイバー複合材料に含まれる各成分について詳細に説明する。
【0016】
<(A)硬化性樹脂組成物>
(a−1)チオール基を有するシルセスキオキサン化合物
本発明で用いられる硬化性樹脂組成物(以下、単に(A)成分とも称する)に含まれるチオール基を有するシルセスキオキサン化合物(以下、単に成分(a−1)とも称する)は、下記化学式(1)で表されるチオール基含有アルコキシシラン類(a−11)を加水分解および縮合して得られる化合物である。
【0017】
【化1】
【0018】
前記化学式(1)中、R1は少なくとも1つのチオール基を有する炭素数1〜8の炭化水素基、または少なくとも1つのチオール基を有する芳香族炭化水素基を表し、R2は水素原子、炭素数1〜8の炭化水素基、または芳香族炭化水素基を表す。
【0019】
上記チオール基含有アルコキシシラン類(a−11)(以下、単に(a−11)成分とも称する)の具体例としては、例えば、3−メルカプトプロピルトリメトキシシラン、3−メルカプトプロピルトリエトキシシラン、3−メルカプトプロピルトリプロポキシシラン、3−メルカプトプロピルトリブトキシシラン、1,4−ジメルカプト−2−(トリメトキシシリル)ブタン、1,4−ジメルカプト−2−(トリエトキシシリル)ブタン、1,4−ジメルカプト−2−(トリプロポキシシリル)ブタン、1,4−ジメルカプト−2−(トリブトキシシリル)ブタン、2−メルカプトメチル−3−メルカプトプロピルトリメトキシシラン、2−メルカプトメチル−3−メルカプトプロピルトリエトキシシラン、2−メルカプトメチル−3−メルカプトプロピルトリプロポキシシラン、2−メルカプトメチル−3−メルカプトプロピルトリブトキシシラン、1,2−ジメルカプトエチルトリメトキシシラン、1,2−ジメルカプトエチルトリエトキシシラン、1,2−ジメルカプトエチルトリプロポキシシラン、1,2−ジメルカプトエチルトリブトキシシラン等が挙げられる。これらは単独でも、また2種以上組み合わせても用いることができる。これら(a−11)成分のうち、3−メルカプトプロピルトリメトキシシランは、加水分解反応の反応性が高く、かつ入手が容易であるため好ましい。
【0020】
また、上記(a−11)成分に加えて、トリメチルメトキシシラン、トリメチルエトキシシラン、トリエチルメトキシシラン、トリエチルエトキシシラン、トリフェニルメトキシシラン、トリフェニルエトキシシランなどのトリアルキルアルコキシシラン類、ジメチルジメトキシシラン、ジメチルジエトキシシラン、ジエチルジメトキシシラン、ジエチルジエトキシシラン、ジフェニルジメトキシシラン、ジフェニルジエトキシシラン、メチルフェニルジメトキシシラン、メチルフェニルジエトキシシラン、3−メルカプトプロピルメチルジメトキシシランなどのジアルキルジアルコキシシラン類、メチルトリメトキシシラン、メチルトリエトキシシラン、エチルトリメトキシシラン、エチルトリエトキシシラン、フェニルトリメトキシシラン、フェニルトリエトキシシランなどのアルキルトリアルコキシシラン類、テトラメトキシシラン、テトラエトキシシラン、テトラプロポキシシラン、テトラブトキシシランなどのテトラアルコキシシラン類、テトラメトキシチタン、テトラエトキシチタン、テトラプロポキシチタン、テトラブトキシチタンなどのテトラアルコキシチタン類、テトラエトキシジルコニウム、テトラプロポキシジルコニウム、テトラブトキシジルコニウムなどのテトラアルコキシジルコニウム類などの金属アルコキシド類(a−12)(以下、単に(a−12)成分とも称する)を使用することができる。これら(a−12)成分は、単独でも、または2種以上組み合わせても用いることができる。これらのうち、トリアルキルアルコキシシラン類、ジアルキルジアルコキシシラン類、テトラアルコキシシラン類を用いることで、(A)成分の架橋密度を調整することができる。アルキルトリアルコキシシラン類を用いることで、(A)成分中に含まれるチオール基の量を調整することができる。
【0021】
(a−11)成分と(a−12)成分とを併用する場合は、{(a−11)成分に含まれるチオール基のモル数}/{(a−11)成分と(a−12)成分との合計モル数}(1分子あたりに含まれるチオール基の平均個数を示す)が0.2以上であることが好ましい。0.2未満である場合、得られる(a−1)成分中に含まれるチオール基の数が少なくなるため、熱硬化性および紫外線硬化性が低下する傾向がある。また、{(a−11)成分と(a−12)成分とに含まれる各アルコキシ基の合計モル数}/{(a−11)成分と(a−12)成分との合計モル数}(モル比:1分子あたりに含まれるアルコキシ基の平均個数を示す)が2.5以上3.5以下であることが好ましく、2.7以上3.2以下であることがより好ましい。2.5未満の場合、得られる(a−1)成分の架橋密度が低く、複合材料の耐熱性が低下する傾向がある。また、3.5を超える場合、(a−1)成分を製造する際、ゲル化しやすくなる傾向がある。
【0022】
本発明に用いられる(a−1)成分は、(a−11)成分単独やこれに(a−12)成分を併用して、それらを加水分解後、縮合させて得ることができる。加水分解反応によって、(a−11)成分や(a−12)成分に含まれるアルコキシ基が水酸基となり、アルコールが副生する。加水分解反応に必要な水の量は、{加水分解反応に用いる水のモル数}/{(a−11)成分と(a−12)成分とに含まれる各アルコキシ基の合計モル数}(モル比)が0.4以上10以下であればよく、好ましくは0.5以上2以下である。0.4未満の場合、(a−1)成分中に加水分解されずにアルコキシ基が残る虞がある。また、10を超える場合、後に行う縮合反応(脱水反応)の際に除くべき水の量が多くなるため製造時間が長くなり、経済的に不利になる傾向がある。
【0023】
また、(a−12)成分としてテトラアルコキシチタン類やテトラアルコキシジルコニウム類等、特に加水分解性および縮合反応性の高い金属アルコキシド類を併用する場合には、急速に加水分解および縮合反応が進行し、系がゲル化してしまう場合がある。この場合、(a−11)成分の加水分解反応を終了させ、実質的にすべての水が消費された状態にした後、該(a−12)成分を添加することによって、ゲル化を避けることができる。
【0024】
加水分解反応に用いる触媒としては、特に限定されず、従来公知の加水分解触媒を任意に用いることができる。これらのうちギ酸は、触媒活性が高く、また引き続く縮合反応の触媒としても機能するので好ましい。ギ酸の添加量は、(a−11)成分および(a−12)成分の合計100質量部に対して、0.1〜25質量部であることが好ましく、1〜10質量部であることがより好ましい。25質量部よりも多いと、(A)成分の安定性が低下する傾向があり、また後工程でギ酸を除去できるとしても該除去量が多くなる傾向がある。一方、0.1質量部よりも少ないと、実質的に反応が進行しない、または反応時間が長くなるなどの傾向がある。反応温度、反応時間は、(a−11)成分や(a−12)成分の反応性に応じて任意に設定できるが、通常0〜100℃程度、好ましくは20〜60℃、1分〜2時間程度である。該加水分解反応は、溶剤の存在下または不存在下に行うことができる。溶剤の種類は特に限定されず、任意の溶剤を1種類以上選択して用いることができるが、後述の縮合反応に用いる溶剤と同一のものを用いることが好ましい。(a−11)成分や(a−12)成分の反応性が低い場合は、無溶剤で行うことが好ましい。
【0025】
該溶剤の例としては、例えば、トルエン、キシレン、メタノール、エタノール、水、プロピレングリコールモノメチルエーテルアセテート、エチレングリコールジメチルエーテル、トルエン等が挙げられる。
【0026】
上記方法で加水分解反応を行うが、{加水分解されてできた水酸基のモル数}/{(a−11)成分と(a−12)成分とに含まれる各アルコキシ基の合計モル数}(モル比)が0.5以上になるように進行させることが好ましく、0.8以上になるように進行させることがさらに好ましい。
【0027】
縮合反応においては、前記の水酸基間で水が副生し、また水酸基とアルコキシ基との間ではアルコールが副生して、シロキサン結合を生じる。縮合反応には、従来公知の脱水縮合触媒を任意に用いることができる。前記のように、ギ酸は触媒活性が高く、加水分解反応の触媒と共用できるため好ましい。反応温度、反応時間は(a−11)成分や(a−12)成分の反応性に応じてそれぞれ任意に設定できるが、通常は40〜150℃程度、好ましくは60〜100℃、30分〜12時間程度である。
【0028】
上記方法で縮合反応を行うが、{未反応の水酸基および未反応のアルコキシ基の合計モル数}/{(a−11)成分と(a−12)成分とに含まれる各アルコキシ基の合計モル数}(モル比)が0.3以下になるように進行させることが好ましく、0.2以下になるように進行させることがより好ましい。0.3を超える場合、未反応の水酸基およびアルコキシ基が(A)成分の保管中に縮合反応してゲル化したり、硬化後に縮合反応し揮発分が発生してクラックが発生するなど、複合材料の性能を損なう傾向がある。
【0029】
当該縮合反応は、(a−11)成分((a−12)成分を併用する場合は両者)の濃度が2〜80質量%程度になるように溶剤希釈して行うことが好ましく、15〜60質量%であることがより好ましい。縮合反応によって生成する水およびアルコールの沸点より高い沸点を有する溶剤を用いると、反応系中よりこれらを留去することができるため好ましい。該濃度が2質量%未満である場合は、(A)成分に含まれる(a−1)成分が少なくなる傾向がある。80質量%を超える場合は、反応中にゲル化したり、生成する(A)成分の分子量が大きくなり過ぎ、複合材料の保存安定性が悪くなる傾向がある。溶剤としては、任意の溶剤を1種類以上選択して用いることができる。縮合反応によって生成する水およびアルコールより高い沸点を有する溶剤を用いれば、反応系中よりこれらを留去することができるため好ましい。また、後述のエチレン性不飽和結合を有する化合物も溶剤の一部として用いることができる。
【0030】
当該縮合反応の終了後、用いた触媒を除去すると、最終的に得られる複合材料の安定性が向上するため好ましい。除去方法は、用いた触媒に応じて公知各種の方法から適宜に選択できる。例えば、ギ酸を用いた場合は、縮合反応の終了後、該沸点以上に加熱する、減圧するなどの方法により容易に除去でき、この点からもギ酸の使用が好ましい。
【0031】
なお、シルセスキオキサン化合物(a−1)は、ランダム構造、ラダー構造、かご構造、不完全縮合かご構造などの構造を有しうることが知られているが、本発明においては、いずれの構造であっても用いることができる。
【0032】
(a−2)エチレン性不飽和結合を有する化合物またはイソシアネート基を有する化合物
本発明で用いられるエチレン性不飽和結合を有する化合物(以下、単に(a−21)成分とも称する)またはイソシアネート基を有する化合物(以下、単に(a−22)成分とも称する)は、(a−1)成分の硬化剤としての役割を果たす。
【0033】
(a−21)成分中のエチレン性不飽和結合は、エチレン性不飽和結合を有する官能基とチオール基との反応より優先して、エチレン性不飽和結合を有する官能基同士が重合する不都合が起こらないよう、ラジカル重合性が低いものを用いることが好ましい。このような(a−21)成分として、アリル基を1つ以上有する化合物が挙げられる。アリル基を1つ含有する化合物としては、例えば、ケイ皮酸、モノアリルシアヌレート、モノアリルイソシアヌレート、ペンタエリスリトールモノアリルエーテル、トリメチロールプロパンモノアリルエーテル、グリセリンモノアリルエーテル、ビスフェノールAモノアリルエーテル、ビスフェノールFモノアリルエーテル、エチレングリコールモノアリルエーテル、ジエチレングリコールモノアリルエーテル、トリエチレングリコールモノアリルエーテル、プロピレングリコールモノアリルエーテル、ジプロピレングリコールモノアリルエーテル、トリプロピレングリコールモノアリルエーテルなどが挙げられる。アリル基を2つ含有する化合物としては、例えば、ジアリルフタレート、ジアリルイソフタレート、ジアリルシアヌレート、ジアリルイソシアヌレート、ペンタエリスリトールジアリルエーテル、トリメチロールプロパンジアリルエーテル、グリセリンジアリルエーテル、ビスフェノールAジアリルエーテル、ビスフェノールFジアリルエーテル、エチレングリコールジアリルエーテル、ジエチレングリコールジアリルエーテル、トリエチレングリコールジアリルエーテル、プロピレングリコールジアリルエーテル、ジプロピレングリコールジアリルエーテル、トリプロピレングリコールジアリルエーテルなどが挙げられる。アリル基を3つ以上含有する化合物としては、例えば、トリアリルイソシアヌレート、ペンタエリスリトールトリアリルエーテル、ペンタエリスリトールテトラアリルエーテル、トリメチロールプロパントリアリルエーテルなどが挙げられる。これら(a−21)成分は、単独でも、または2種以上組み合わせても用いることができる。これらの中でも、分子中に1つのエチレン性不飽和結合のみを有する化合物では分子間の架橋が起こらないため、複合材料の耐熱性、表面硬度等の物性についての改善効果も不充分となる傾向があることから、分子中に2以上のエチレン性不飽和結合を有する化合物が好ましく、中でもトリアリルイソシアヌレート、ジアリルフタレート、ペンタエリスリトールトリアリルエーテルが特に好ましい。
【0034】
また、(a−21)成分として、上記アリル基を有する化合物よりも高分子量のものを用いることもできる。(a−21)成分として高分子量のものを用いた複合材料は、可撓性が向上する傾向がある。また、一般にラジカル重合性が低くなる傾向があり、このような視点からも好ましく用いることができる。該高分子量物としては、メチルアリルシロキサンとジメチルシロキサンとからなる共重合物、エピクロルヒドリンとアリルグリシジルエーテルとからなる共重合物(ダイソー株式会社製:商品名「エピクロマー(登録商標)」、日本ゼオン株式会社製:商品名「Gechron(登録商標)」など)、アリル基末端ポリイソブチレンポリマー(株式会社カネカ製:商品名「エピオン(登録商標)」)、ウレタンアクリレート(荒川化学工業株式会社製:商品名「ビームセット550B」)などが挙げられる。これらの化合物は、単独でも、または2種以上組み合わせても用いることができる。
【0035】
また、本発明で用いられる(a−22)成分は、特に限定されず、従来公知のイソシアネート基を有する化合物を適宜に用いることができる。該イソシアネート基を有する化合物としては、例えば、芳香族、脂肪族または脂環族の各種公知のジイソシアネート類を使用することができ、より具体的には、例えば、1,5−ナフチレンジイソシアネート、4,4’−ジフェニルメタンジイソシアネート、4,4’−ジフェニルジメチルメタンジイソシアネート、4,4’−ジベンジルイソシアネート、ジアルキルジフェニルメタンジイソシアネート、テトラアルキルジフェニルメタンジイソシアネート、1,3−フェニレンジイソシアネート、1,4−フェニレンジイソシアネート、トリレンジイソシアネート、ブタン−1,4−ジイソシアネート、ヘキサメチレンジイソシアネート、イソプロピレンジイソシアネート、メチレンジイソシアネート、2,2,4−トリメチルヘキサメチレンジイソシアネート、2,4,4−トリメチルヘキサメチレンジイソシアネート、シクロヘキサン−1,4−ジイソシアネート、キシリレンジイソシアネート、水素化キシリレンジイソシアネート、イソホロンジイソシアネート、リジンジイソシアネート、ジシクロヘキシルメタン−4,4’−ジイソシアネート、1,3−ビス(イソシアネートメチル)シクロヘキサン、メチルシクロヘキサンジイソシアネート、m−テトラメチルキシリレンジイソシアネートやダイマー酸のカルボキシル基をイソシアネート基に転化したダイマージイソシアネートなどが挙げられる。これらの化合物は、単独でも、または2種以上組み合わせても用いることができる。該化合物のうち、イソホロンジイソシアネートは、最終的に得られる複合材料が透明性、耐熱性等に優れ、かつ入手が容易であるため特に好ましい。
【0036】
また、(a−22)成分として、前記化合物よりも高分子量のものを用いることができる。高分子量のものを用いてなる複合材料は、可撓性が向上する傾向がある。該高分子量物としては、ポリカーボネートジオール、ポリエステルジオールなどのポリオール類のジイソシアネート変性物、ポリメリックMDI(三井武田ケミカル株式会社製:商品名「コスモネート(登録商標)M」など)、ポリイソシアヌレートタイプのHDI(日本ポリウレタン工業株式会社製:商品名「コロネート(登録商標)HX」など)などが挙げられる。これらの化合物は、単独でも、または2種以上組み合わせても用いることができる。これら化合物のうち、ポリイソシアヌレートタイプのHDIは、最終的に得られる複合材料が透明性、耐熱性等に優れ、かつ入手が容易であるため好ましい。
【0037】
また、高分子量物としてメタクリロイルオキシエチルイソシアネート(昭和電工株式会社製:商品名「カレンズMOI(登録商標)」)、アクリロイルオキシエチルイソシアネート(昭和電工株式会社製:商品名「カレンズAOI(登録商標)」)など、1分子中にエチレン性不飽和結合およびイソシアネート基が共に存在する化合物を用いることもできる。このような化合物を用いた際には、(a−21)成分および(a−22)成分を同時に含有するものとみなすことができ、分子中に含まれるエチレン性不飽和結合の数およびイソシアネート基の数を考慮のうえ使用量を決定する必要がある。
【0038】
また、(a−22)成分を用いる場合には、従来公知のウレタン化触媒を用いることができる。例えば、ジブチルスズジラウレート、オクチル酸スズなどの有機スズ化合物、1,8−ジアザ−ビシクロ[5.4.0]ウンデセン−7、トリエチレンジアミン、ベンジルジメチルアミン、トリエタノールアミン、ジメチルアミノエタノール、トリス(ジメチルアミノメチル)フェノールなどの三級アミン類などをあげることができる。ウレタン化触媒は、(A)成分100質量部に対し、0.01〜5質量部の割合で使用することが好ましい。
【0039】
上記(A)成分における有効(a−1)成分、(a−21)成分および(a−22)成分の濃度は、用途に応じて適宜に決定でき、必要に応じて溶剤を配合することができる。溶剤としては、当該成分と非反応性であればよく、各種従来公知のものを適宜選択して用いることができる。(A)成分を1mm以上の厚膜に硬化させる場合は、(a−1)成分、(a−21)成分および(a−22)成分の合計濃度を(A)成分中90質量%以上にすることが好ましく、95質量%以上にすることがより好ましい。該合計濃度は、(a−1)成分、(a−21)成分および(a−22)成分の濃度と(A)成分の仕込み時に加えた溶剤の量により計算で求めることができ、また(A)成分に含まれる溶剤の沸点以上で2時間程度加熱し、加熱前後の重量変化により求めることもできる。(A)成分を1mm以上の厚膜に硬化させる場合は、90質量%未満の場合、硬化、成型時に発泡したり、複合材料中に溶剤が残存したりして、複合材料の物性が低下する傾向がある。なお、(a−1)成分を合成する際に溶剤を使用しているため、反応終了後、不揮発分含有量が90質量%以上となるよう溶剤を揮発させておけばよい。また、(A)成分を調製した後、用いた溶剤を揮発させて、有効な(a−1)成分、(a−21)成分および(a−22)成分の合計濃度を高めることもできる。
【0040】
上記(A)成分は、(a−1)成分、ならびに(a−21)成分および(a−22)成分の少なくとも一方を含有するものである。(A)成分中に含有される各成分は、[{(a−21)成分中に含まれるエチレン性不飽和結合の数}/{(a−1)成分中のチオール基の数}]が0.1〜0.8、[{(a−22)成分中のイソシアネート基の数}/{(a−1)成分中のチオール基の数}]が0.1〜0.8、[{(a−21)成分中に含まれるエチレン性不飽和結合の数+(a−22)成分中に含まれるイソシアネート基の数}/{(A)成分中のチオール基の数}]が0.9〜1.1なる割合で含有することが好ましい。
【0041】
[{(a−21)成分中に含まれるエチレン性不飽和結合の数+(a−22)成分中に含まれるイソシアネート基の数)}/{(a−1)成分中のチオール基の数}]が0.9未満の場合には、チオール基が残存し、その分解によって悪臭を発生させる場合がある。1.1を超える場合には、硬化後にエチレン性不飽和結合やイソシアネート基が残存し、耐候性が低下する傾向がある。
【0042】
[{(a−21)成分中に含まれるエチレン性不飽和結合の数}/{(a−1)成分中のチオール基の数}]が0.8を超えると、紫外線硬化を第一段階とする場合には硬化が進行しすぎ、熱硬化を第一段階とする場合には半硬化物中に未反応の(a−21)成分が多くなりすぎるため、いずれも成型加工性が失われる場合がある。また、0.1未満の場合は、成型加工性が失われる場合がある。
【0043】
{(a−22)成分中のイソシアネート基の数}/{(a−1)成分中のチオール基の数)}が0.8を超えると、紫外線硬化を第一段階とする場合には半硬化物中に未反応の(a−22)成分が多くなりすぎるため、熱硬化を第一段階とする場合には硬化が進行しすぎ、いずれも成形加工性が失われる場合がある。また、0.1未満の場合は、成形加工性が失われる場合がある。
【0044】
また、(A)成分には、用途に応じ、上記(a−11)成分および/またはその加水分解物(以下、併せて(E)成分とも称する)を配合できる。(E)成分は、(a−1)成分の合成に際して用いた(a−11)成分をそのままで用いるか、その加水分解物を用いるか、これらを組み合わせて使用できる。(E)成分の配合量は、(A)成分100質量部に対して、0.1〜20質量部程度であることが好ましい。0.1質量部未満の場合は、複合材料の無機基材に対する密着性向上効果が不充分となる傾向がある。また、20質量部を超える場合、(E)成分が加水分解、縮合反応する際の揮発分が多くなるため、(A)成分が硬化時に発泡したり、反りやクラックが発生したり、得られる複合材料が脆くなったりする傾向がある。このような(E)成分としては、3−メルカプトプロピルトリメトキシシランが、密着性向上効果の点で特に好ましい。
【0045】
さらに、(A)成分には、用途に応じ、上記(a−12)成分である金属アルコキシド類および/またはその加水分解物(以下、併せて(F)成分とも称する)を配合できる。(F)成分は、(a−1)成分の合成に際して用いた金属アルコキシド類をそのままで用いるか、その加水分解物を用いるか、これらを組み合わせて使用できる。(F)成分を含有する(A)成分を用いることで、得られる複合材料の屈折率を調整することができる。(F)成分の配合量は、(A)成分100質量部に対して、0.1〜20質量部程度であることが好ましい。0.1質量部に満たない場合には、屈折率向上効果が不充分となる傾向がある。また、20質量部を超える場合は、(F)成分が加水分解、縮合反応する際の揮発分が多くなるため、(A)成分が硬化時に発泡したり、反りやクラックが発生したり、得られる複合材料が脆くなったりする傾向がある。
【0046】
(A)成分は、熱硬化用触媒および/または光硬化用触媒を含むことが好ましい。前記熱硬化用触媒の例としては、従来公知のウレタン化触媒を用いることができる。例えば、ジブチル錫ジラウリレート、オクチル酸錫などの有機錫化合物、1,8−ジアザ−ビシクロ[5.4.0]ウンデセン−7、トリエチルジアミン、ベンジルメチルアミン、トリエタノールアミン、ジメチルアミノエタノール、トリス(ジメチルアミノメチル)フェノールなどの三級アミン類などをあげることができる。また、前記光硬化用触媒の例としては、従来公知の光カチオン開始剤、光ラジカル開始剤などを任意に選択できる。光カチオン開始剤としては、紫外線の照射により酸を発生する化合物であるスルホニウム塩、ヨードニウム塩、メタロセン化合物、ベンゾイントシレート等があげられ、それらの市販品としては、例えば、サイラキュア(登録商標)UVI−6970、同UVI−6974、同UVI−6990(いずれも米国ユニオンカーバイド社製商品名)、イルガキュア(登録商標)264(チバスペシャルティケミカルズ社製)、CIT−1682(日本曹達株式会社などが挙げられる。光ラジカル開始剤としては、ダロキュア(登録商標)1173、イルガキュア(登録商標)651、イルガキュア(登録商標)184、イルガキュア(登録商標)907(いずれもチバスペシャルティケミカルズ社製商品名)、ベンゾフェノン等が挙げられる。なお、得られる硬化物の耐候性低下が懸念される場合、特に高い耐候性、透明性が求められる光学部材などに用いられる場合には、光反応開始剤や光増感剤を使用しないほうがよい。
【0047】
また、硬化性樹脂組成物(A)の安定性をより向上させるため、エン−チオール反応を抑制する化合物を配合できる。このような化合物としては、トリフェニルホスフィン、亜リン酸トリフェニル等のリン系化合物;p−メトキシフェノール、ハイドロキノン、ピロガロ−ル、ナフチルアミン、tert−ブチルカテコール、塩化第一銅、2,6−ジ−tert−ブチル−p−クレゾール、2,2’−メチレンビス(4−エチル−6−tert−ブチルフェノール)、2,2’−メチレンビス(4−メチル−6−tert−ブチルフェノール)、N−ニトロソフェニルヒドロキシルアミンアルミニウム塩、ジフェニルニトロソアミン等のラジカル重合禁止剤;ベンジルジメチルアミン、2−(ジメチルアミノメチル)フェノール、2,4,6−トリス(ジアミノメチル)フェノール、ジアザビシクロウンデセン等の3級アミン類;2−メチルイミダゾール、2−エチル−4−メチルイミダゾール、2−エチルへキシルイミダゾール、2−ウンデシルイミダゾール、1−シアノエチル−2−メチルイミダゾール等のイミダゾール類があげられる。
【0048】
(A)成分には、本発明の効果を損なわない範囲で、各種用途での必要性に応じて、可塑剤、耐候剤、酸化防止剤、熱安定剤、滑剤、帯電防止剤、増白剤、着色剤、導電剤、離型剤、表面処理剤、粘度調節剤、フィラー等を配合してもよい。
【0049】
<硬化性樹脂組成物の製造方法>
本発明に用いられる硬化性樹脂組成物の製造方法は、特に制限されないが、上記の(a−1)成分、(a−2)成分、ならびに必要に応じて熱硬化触媒、光硬化触媒、および添加剤などを、溶媒中で一括に混合するか、各成分を順次混合するか、または任意の複数の成分を混合した後に残りの成分を混合するなどして、均一な混合物となるように攪拌する方法が挙げられる。
【0050】
調製に用いる溶媒としては、例えば、エチレングリコールジメチルエーテル、トルエン等が挙げられる。
【0051】
(A)成分の調製は、例えば、スターラーなどで均一になるまで、10〜50℃の温度範囲で、5〜60分攪拌することにより行うことができる。
【0052】
このようにして得られる硬化性樹脂組成物(A)の硬化後の屈折率は、1.53〜1.57である。屈折率が1.53未満の場合には、キチンナノファイバー不織布との屈折率の違いが大きく、最終的に得られる複合材料のヘイズが低下する。一方、1.57を超える場合には、複屈折が生じ、最終的に得られる複合材料のヘイズが低下する。該屈折率は、好ましくは1.532〜1.565である。
【0053】
該屈折率は、硬化性樹脂組成物(A)に含まれる各成分の配合比を調整することにより、所望の値とすることができる。また、該屈折率は、実施例に記載の方法により測定することができる。
【0054】
<キチンナノファイバー不織布>
本発明で用いられるキチンナノファイバー不織布は、平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーを絡合して得られるものである。
【0055】
前記平均繊維径が45nm未満であると、キチンナノファイバー不織布に硬化性樹脂組成物を空隙なく含浸させることが困難となり、空隙による光散乱が生じ、その結果複合材料のヘイズが悪化する。一方、65μmを超えると、キチンナノファイバーによる補強効果が薄れ、複合材料の耐熱性および寸法安定性が悪化する。繊維径分布が1.3を超えると、繊維径の大きな部分での光散乱が生じ、その結果複合材料のヘイズが悪化する。該平均繊維径は好ましくは41〜48nmであり、該繊維径分布は好ましくは1.1〜1.28である。なお、繊維径分布は、定義上、1未満の値にはならない。該平均繊維径および該繊維径分布は、実施例に記載の方法により測定することができる。
【0056】
上記のような平均繊維径および繊維径分布を有するキチンナノファイバーを得る方法は特に制限されないが、例えば、市販の精製キチンを酸性液体に浸漬させた後、解繊処理を行う方法が挙げられる。
【0057】
前記酸性液体としては、所望の範囲のpHが得られる限度で任意の酸を用いることができる。すなわち、酸は、有機酸であってもよく、無機酸であってもよく、特に制限されない。また、酸性液体の溶媒にも特に限定はなく、水以外のものを用いてもよい。
【0058】
前記有機酸としては、例えば、ギ酸、酢酸、クエン酸、リンゴ酸、シュウ酸、サリチル酸、アスコルビン酸、酒石酸、グルコン酸、乳酸、フマル酸、コハク酸、コハク酸ナトリウム、フィチン酸、アジピン酸、プロピオン酸、グリオキシル酸、ピルビン酸、アセト酢酸、レブリン酸、ヘプタン酸、カプリル酸、カプリン酸、ラウリル酸、グリコール酸、グリセリン酸、アクリル酸、安息香酸、パラニトロ安息香酸、パラトルエンスルホン酸、ピクリン酸、マレイン酸などが挙げられる。前記無機酸としては、例えば、リン酸、塩酸、硫酸、硝酸、ピロリン酸二水素二ナトリウムなどが挙げられる。
【0059】
前記酸性液体のpHは5以下であることが好ましい。かような範囲であれば、上記範囲の平均繊維径および繊維径分布を有するキチンナノファイバーを効率よく得ることができる。
【0060】
前記解繊処理は、例えば、家庭用ミキサー(プロペラミキサー、カッターミキサー)、超音波ホモジナイザー、高圧ホモジナイザー、二軸混練機、石臼式粉砕機などの解繊・粉砕装置を用いて行うことができる。
【0061】
上記方法によりキチンナノファイバーを得た後、絡合処理を行うことにより、キチンナノファイバー不織布を得ることができる。絡合処理の方法も特に制限されず、乾式法や湿式法など、いずれの方法で行ってもよい。
【0062】
得られたキチンナノファイバー不織布に、上記硬化性樹脂組成物を含浸させプリプレグを作製し、該プリプレグを硬化させることにより、本発明の複合材料が得られる。プリプレグを得る方法、およびプリプレグを硬化させる方法については、後述する。
【0063】
<キチンナノファイバー複合材料の製造方法>
本発明のキチンナノファイバー複合材料の製造方法は特に制限されないが、下記の工程1〜4を含むことが好ましい。
【0064】
(工程1)チオール基を有するシルセスキオキサン化合物(a−1)とエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)とを混合し、屈折率が1.53〜1.57である硬化性樹脂組成物(A)を得る工程。
【0065】
(工程2)平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーを絡合させてキチンナノファイバー不織布(B)を得る工程。
【0066】
(工程3)前記硬化性樹脂組成物(A)を前記キチンナノファイバー不織布(B)に含浸させてプリプレグを得る工程。
【0067】
(工程4)前記プリプレグを硬化させる工程。
【0068】
これらの工程の中でも、工程1および工程2は、上記で説明した通りであるため、ここでは説明を省略する。以下では、工程3および工程4について説明する。
【0069】
(工程3)
本工程では、硬化性樹脂組成物(A)をキチンナノファイバー不織布(B)に含浸させてプリプレグを作製する。
【0070】
硬化性樹脂組成物(A)をキチンナノファイバー不織布(B)に含浸させる方法は特に制限されないが、例えば、浸漬などによりキチンナノファイバー不織布(B)内部に均一に含浸させる方法、キチンナノファイバー不織布(B)の表面と裏面とに硬化性樹脂組成物(A)を塗布する方法などが挙げられる。
【0071】
キチンナノファイバー不織布(B)に対する硬化性樹脂組成物(A)の使用割合は、特に制限されないが、通常はキチンナノファイバー不織布(B)100質量部あたり20〜500質量部程度である。
【0072】
(工程4)
本工程では、工程3で得られたプリプレグを硬化させ、キチンナノファイバー複合材料を得る。
【0073】
硬化の前に、プリプレグを加熱して溶剤乾燥を行うことが好ましい。加熱乾燥の条件は特に制限されないが、通常70〜100℃の温度で、2〜15分間行われる。
【0074】
加熱して溶剤を乾燥させた後、熱硬化および紫外線硬化の少なくとも一方を行う。
【0075】
前記熱硬化は、90〜130℃の温度で、5〜30分間行うことが好ましい。また、紫外線硬化の際の積算光量は250〜2000mJ/cm2であることが好ましい。
【0076】
熱硬化および紫外線硬化の両方を行う場合は、一段階目で熱硬化を行った後二段階目で紫外線硬化を行ってもよいし、一段階目で紫外線硬化を行った後二段階目で熱硬化を行ってもよい。
【0077】
上記硬化が終了した後は、必要に応じてアニール処理を行う。アニール処理の条件も特に制限されず、通常100〜180℃の温度範囲で、10〜60分間加熱処理することにより行われる。
【0078】
こうして得られる本発明のキチンナノファイバー複合材料は、例えば、液晶パネルや有機ELディスプレイなどのディスプレイ用基板、導光板、偏光板、PDPパネル、OHPフィルム、光ファイバー、カラーフィルター、光ディスク基板、レンズ、液晶セル用プラスチック基板などに好適に用いられる。
【実施例】
【0079】
本発明を、以下の実施例および比較例を用いてさらに詳細に説明する。ただし、本発明の技術的範囲が以下の実施例のみに制限されるわけではない。なお、屈折率、平均繊維径、および繊維径分布の測定は、以下の方法で行った。
【0080】
・屈折率:株式会社アタゴ製、アッベ屈折率計 DR−M2を用いて、波長589nmの屈折率を測定した。
【0081】
・平均繊維径および繊維径分布:ナノファイバー不織布を走査型電子顕微鏡で観察、写真撮影を行い、得られた写真に直線を数本引き、この線を横切る繊維の直径を1枚の写真から200本読み取る。これを、それぞれのナノファイバー不織布について5回繰り返し、合計で1,000本の繊維径のデータから平均繊維径および繊維径分布を求める。それぞれの算出方法は、次の式に従った。
【0082】
【数1】
【0083】
(a−1)チオール基を有するシルセスキオキサン化合物
(a−1−1)の合成
攪拌機、冷却管、分水器、温度計、窒素吹き込み口を備えた反応装置に、3−メルカプトプロピルトリメトキシシラン(信越化学工業株式会社製、商品名「KBM−803」)3400質量部、イオン交換水936質量部、95%ギ酸68質量部を仕込み、室温(25℃)で30分間加水分解反応させた。反応中、発熱によって最大35℃温度上昇した。反応後、トルエン5670質量部を仕込み、加熱した。71℃まで昇温したところで、加水分解によって発生したメタノールと、トルエンの一部が留去され始めた。2時間かけて75℃まで昇温し、縮合反応させて水を留去した。さらに1時間、75℃で反応させた後、70℃、200hPaで減圧して、残存するトルエンの一部、メタノール、水、およびギ酸を留去した。さらに70℃、7hPaで減圧してトルエンを留去することで、チオール基を含有するシルセスキオキサン化合物(a−1−1)を2330質量部得た。濃度は99.0%であった。また、(a−1−1)のチオール基の濃度は、7.41ミリモル/gであった。
【0084】
(a−1−2)の合成
(a−1−1)の合成で用いた装置と同様の装置に、3−メルカプトプロピルトリメトキシシラン15.0質量部、フェニルトリメトキシシラン(東京化成工業株式会社製)5.05質量部、イオン交換水5.51質量部、95%ギ酸1.00質部を仕込み、室温(25℃)で30分間加水分解反応させた。反応中、発熱によって最大20℃温度上昇した。反応後、プロピレングリコールモノメチルエーテルアセテート19.52質量部を仕込み、加熱した。82℃まで昇温したところで、加水分解によって発生したメタノールが留去され始めた。30分かけて105℃まで昇温し、縮合反応させて水を留去した。さらに1時間30分、105℃で反応させた後、70℃、20kPa(150mmHg)で減圧して、残存するメタノール、水、ギ酸を留去することで、チオール基を含有するシルセスキオキサン化合物(a−1−2)を25.13質量部得た。濃度は51.8%であった。また、縮合物(a−1−2)のチオール当量は、329g/eqであった。
【0085】
(a−1−3)の合成
(a−1−1)の合成で用いた装置と同様の装置に、3−メルカプトプロピルトリメトキシシラン180質量部、イオン交換水49.55質量部、95%ギ酸9.00質量部を仕込み、室温(25℃)で30分間加水分解反応させた。反応中、発熱によって最大22℃温度上昇した。反応後、トルエン272.23質量部を仕込み、加熱した。72℃まで昇温したところで、加水分解によって発生したメタノールとトルエンの一部が留去され始めた。20分かけて75℃まで昇温し、縮合反応させて水を留去した。さらに1時間、75℃で反応させた後、70℃、20kPa(150mmHg)で減圧して、残存するメタノール、水、ギ酸を留去した。さらに70℃、0.67kPa(5mmHg)で減圧して、トルエンを留去することで、チオール基を含有するシルセスキオキサン化合物(a−1−3)を124.49質量部得た。濃度は93.7%であった。また、シルセスキオキサン化合物(a−1−3)のチオール当量は、136g/eqであった。
【0086】
(a−1−4)の合成
(a−1−1)の合成で用いた装置と同様の装置に、3−メルカプトプロピルトリメトキシシラン(東レ・ダウコーニング株式会社製、商品名「SH−6062」)190質量部、イオン交換水52.3質量部、95%ギ酸9.5質量部を仕込み、室温(25℃)で30分間加水分解反応させた。反応中、発熱によって最大22℃温度上昇した。反応後、プロピレングリコールモノメチルエーテルアセテート(日本乳化剤株式会社製、商品名「MFG−AC」)287.36質量部を仕込み、加熱した。82℃まで昇温したところで、加水分解によって発生したメタノールが留去され始めた。30分かけて105℃まで昇温し、縮合反応によって発生した水を留去した。さらに1時間30分、105℃で反応させた後、70℃、20kPa(150mmHg)で減圧して、残存するメタノール、水、ギ酸、およびプロピレングリコールモノメチルエーテルアセテートの一部を留去することで、チオール基を含有するシルセスキオキサン化合物(a−1−4)を385.2g得た。濃度は32.0%であった。また、(a−1−4)のチオール当量は、398g/eqであった。
【0087】
(A)硬化性樹脂組成物の製造
(A−1)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−1)31.7質量部に対して、硬化剤である(a−2)成分としてトリアリルイソシアヌレート(以下TAICとも称する、日本化成株式会社製、商品名「TAIC」、エチレン性不飽和結合の濃度は12.0ミリモル/g)5.4質量部、および多官能イソシアネート(日本ポリウレタン株式会社製、商品名「コロネート(登録商標)HX」、イソシアネート基の濃度は5.00ミリモル/g)33.0質量部、熱硬化用触媒としてジブチルスズジラウレート(以下U−100とも称する、日東化成株式会社製、商品名「ネオスタンU−100」)0.15質量部、光硬化用触媒として1−ヒドロキシシクロヘキシルフェニルケトン(以下Irg184とも称する、チバスペシャルティケミカルズ株式会社製、商品名「イルガキュア(登録商標)184」)0.15質量部、および希釈溶剤としてエチレングリコールジメチルエーテル29.6質量部を配合した。25℃で15分間攪拌混合し、硬化性樹脂組成物(A−1)を得た。
【0088】
(A−2)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−2)23.9質量部に対し、硬化剤である(a−2)成分としてTAIC6.0質量部、亜リン酸トリフェニル(東京化成工業株式会社製)0.4質量部、および希釈溶剤としてエチレングリコールジメチルエーテル69.7質量部を配合し、25℃で15分間攪拌混合し、硬化性樹脂組成物(A−2)を得た。
【0089】
(A−3)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−3)18.6質量部に対し、硬化剤である(a−2)成分としてTAIC11.4質量部、亜リン酸トリフェニル(東京化成工業株式会社製)0.4質量部、および希釈溶剤としてエチレングリコールジメチルエーテル69.6質量部を配合し、25℃で15分間攪拌混合し、硬化性樹脂組成物(A−3)を得た。
【0090】
(A−4)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−1)20質量部に対し、硬化剤である(a−2)成分として多官能ウレタンアクリレート(以下550Bとも称する、荒川化学工業株式会社製、商品名「ビームセット550B」、炭素−炭素二重結合の濃度は2.25ミリモル/g)38.1質量部、多官能イソシアネートとしてコロネート(登録商標)HXを11.8質量部、熱硬化用触媒としてU−100を0.15質量部、光硬化用触媒としてIrg184を0.15質量部、および希釈溶剤としてエチレングリコールジメチルエーテル29.8質量部を配合し、25℃で15分間攪拌混合し、硬化性樹脂組成物(A−4)を得た。
【0091】
(A−5)の製造
チオール基を含有するシルセスキオキサン化合物(a−1−4)23.8質量部に対し、硬化剤である(a−2)成分として550Bを4.9質量部、亜リン酸トリフェニル(東京化成工業株式会社製)0.4質量部、屈折率調整剤としてテトラブトキシチタン1.4質量部、および希釈溶剤としてエチレングリコールジメチルエーテル69.5質量部を配合し、25℃で15分間攪拌混合し、硬化性樹脂組成物(A−5)を得た。
【0092】
(B)キチンナノファイバー不織布
(B−1)の製造
蒸留水100質量部に、精製キチン(ナカライテスク株式会社製)1.5質量部、氷酢酸(キシダ化学株式会社製)0.5質量部を添加し、pHを3に調整した。これを、石臼式粉砕機(増幸産業株式会社製、スーパーマスコロイダー MKCA6−2)で解繊処理を1回行うことによってキチンナノファイバーのペースト状物を得た。このペースト状物100質量部に蒸留水500質量部を加えて希釈液とし、この希釈液を親水性PTFEメンブレンフィルター(日本ミリポア株式会社製、オムニポア 平均空孔径1μm)を用いて吸引濾過することによって、親水性PTFEメンブレンフィルター上にキチンナノファイバー不織布(B−1)を得た。得られたキチンナノファイバー不織布(B−1)の厚みは55μm、空孔率は52%、平均繊維径は48nm、繊維径分布は1.13であった。
【0093】
(B−2)の製造
氷酢酸の量を0.4質量部としpHを4に調整すること以外は、上記(B−1)と同様にして、キチンナノファイバー不織布(B−2)を得た。得られたキチンナノファイバー不織布(B−2)の厚みは52μm、空孔率は49%、平均繊維径は61nm、繊維径分布は1.28であった。
【0094】
(B−3)の製造
氷酢酸の量を0.2質量部としpHを6に調整すること以外は、上記(B−1)と同様にして、キチンナノファイバー不織布(B−3)を得た。得られたキチンナノファイバー不織布(B−3)の厚みは55μm、空孔率は51%、平均繊維径は62nm、繊維径分布は1.35であった。
【0095】
(B−4)の製造
上記(B−1)に記載の1度解繊処理をして得られたキチンナノファイバーペースト状物を、石臼式粉砕機を用いてさらに繰り返し2回解繊処理を加えること以外は、上記(B−1)と同様にして、キチンナノファイバー不織布(B−4)を得た。得られたキチンナノファイバー不織布(B−4)の厚みは48μm、空孔率は47%、平均繊維径は41nm、繊維径分布は1.10であった。
【0096】
(D−1)セルロースナノファイバー不織布の製造
濾紙(アドバンテック社製のFILTER PAPER)をハサミで3mm角に切断したもの2gを200mlのビーカに入れた。そこに、N,N−ジメチルアセトアミド 50mLとイオン液体である塩化1−ブチル−3−メチルイミダゾリウム60gを加え、80℃で磁性攪拌子を用いて60分攪拌した後、6モルの硫酸水溶液9質量部を加え、90℃で60分加水分解反応を行った。その後、反応液を室温(25℃)まで冷却してから100質量部のN,N−ジメチルアセトアミドを投入し、攪拌して分散液とした。その後、遠心分離機を用いて、分散液を蒸留水で置換、洗浄することによって、1−ブチル−3−メチルイミダゾリウム、N−ジメチルアセトアミド、硫酸、および水溶性糖分などを除去したセルロースナノファイバー分散液(固形分濃度0.2質量%)を得た。
【0097】
この分散液を親水性PTFEメンブレンフィルター(日本ミリポア株式会社製、オムニポア 平均空孔径1μm)を用いて吸引濾過することによって、親水性PTFEメンブレンフィルター上にセルロースナノファイバー不織布(D−1)を得た。得られたセルロースナノファイバー不織布(D−1)の厚みは56μm、空孔率は58%、平均繊維径は55nm、繊維径分布は1.45であった。
【0098】
(実施例1)
硬化性樹脂組成物(A−1)をキチンナノファイバー不織布(B−1)に含浸させプリプレグを得た後、PETフィルム上に静置し、ホットプレートを用いて80℃で5分、および110℃で10分の条件で、溶剤乾燥および熱硬化を行った。さらに、紫外線照射装置を用いて254nmの検出器で積算光量が500mJ/cm2となるよう紫外線を照射し紫外線硬化を行った後、120℃のオーブン中で15分間アニールを行い、膜厚75μmの透明なキチンナノファイバー複合材料を得た。なお、(A−1)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.554であった。
【0099】
(実施例2)
硬化性樹脂組成物(A−2)をキチンナノファイバー不織布(B−1)に含浸させプリプレグを得た後、PETフィルム上に静置し、ホットプレートを用いて80℃で5分間溶剤乾燥を行った。次に、紫外線照射装置を用いて254nmの検出器で積算光量が500mJ/cm2となるよう紫外線を照射し紫外線硬化を行った後、120℃のオーブン中で15分間アニールを行い、膜厚73μmの透明なキチンナノファイバー複合材料を得た。なお、(A−2)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.532であった。
【0100】
(実施例3)
硬化性樹脂組成物として(A−3)を使用すること以外は、実施例2と同様の方法により、膜厚78μmの透明なキチンナノファイバー複合材料を得た。なお、(A−3)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.565であった。
【0101】
(比較例1)
硬化性樹脂組成物として(A−4)を使用すること以外は、実施例1と同様の方法により、膜厚76μmの透明なキチンナノファイバー複合材料を得た。なお、(A−4)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.520であった。
【0102】
(比較例2)
硬化性樹脂組成物として(A−5)を使用すること以外は、実施例2と同様の方法により、膜厚72μmの透明なキチンナノファイバー複合材料を得た。なお、(A−5)を(B−1)に含浸せずに単独で硬化させた時の屈折率は1.594であった。
【0103】
(実施例4)
キチンナノファイバー不織布として(B−2)を使用すること以外は、実施例1と同様の方法により、膜厚74μmの透明なキチンナノファイバー複合材料を得た。
【0104】
(比較例3)
キチンナノファイバー不織布として(B−3)を使用すること以外は、実施例1と同様の方法により、膜厚78μmの透明なキチンナノファイバー複合材料を得た。
【0105】
(比較例4)
キチンナノファイバー不織布として(B−4)を使用すること以外は、実施例1と同様な操作を行い膜厚75μmの透明なキチンナノファイバー複合材料を得た。
【0106】
(比較例5)
キチンナノファイバー不織布の代わりにセルロースナノファイバー不織布(D−1)を用いること以外は、実施例1と同様の方法により、膜厚72μmの透明なセルロースナノファイバー複合材料を得た。
【0107】
<評価>
・寸法安定性:株式会社リガク製、TMA8310を用いて、窒素雰囲気下、1分間に5℃の割合で室温(25℃)から250℃まで昇温させ、荷重45mN、引っ張りモードで寸法変化を測定し、27℃から200℃の温度範囲における線膨張係数を算出した。
【0108】
・全光線透過率:株式会社島津製作所製、分光光度計 UV−2200(積分球付き)を用い、380nmから780nmの波長範囲における全光線透過率(Tt)を測定した。
【0109】
・ヘイズ:株式会社島津製作所製、分光光度計 UV−2200(積分球付き)を用い、380nmから780nmの波長範囲における拡散透過率(Td)を測定し、次の式に従いヘイズを算出した。
【0110】
【数2】
【0111】
各実施例および比較例の上記評価方法の結果を下記表1に示す。
【0112】
【表1】
【0113】
上記表1から明らかなように、本発明の複合材料(実施例1〜4)は、本発明の範囲外である複合材料(比較例1〜5)と比べて、寸法安定性およびヘイズに優れる。また、本発明のキチンナノファイバー複合材料は、ガラス材料を含む複合材料に比べて可撓性に優れる。
【0114】
また、実施例1で得られたキチンナノファイバー複合材料および実施例1で用いられた硬化性樹脂組成物の耐熱性を評価した。具体的には、セイコーインスツルメント株式会社製、粘弾性測定器DMS6100を用い、1分間に5℃の割合で室温(25℃)から250℃まで昇温させ、測定周波数1Hzで動的貯蔵弾性率を測定した。その結果を図1に示す。図1から明らかなように、実施例1のキチンナノファイバー複合材料は、硬化性樹脂組成物に比べて高温での弾性率の低下が少なく、耐熱性に優れていることが認められる。
【特許請求の範囲】
【請求項1】
チオール基を有するシルセスキオキサン化合物(a−1)、ならびにエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)を含み、かつ硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)と、
平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーからなるキチンナノファイバー不織布(B)と、
を含むプリプレグを硬化させてなる、キチンナノファイバー複合材料。
【請求項2】
チオール基を有するシルセスキオキサン化合物(a−1)とエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)とを混合し、硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)を得る工程と、
平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーを絡合させてキチンナノファイバー不織布(B)を得る工程と、
前記硬化性樹脂組成物(A)を前記キチンナノファイバー不織布(B)に含浸させてプリプレグを得る工程と、
前記プリプレグを硬化させる工程と、
を含む、キチンナノファイバー複合材料の製造方法。
【請求項1】
チオール基を有するシルセスキオキサン化合物(a−1)、ならびにエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)を含み、かつ硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)と、
平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーからなるキチンナノファイバー不織布(B)と、
を含むプリプレグを硬化させてなる、キチンナノファイバー複合材料。
【請求項2】
チオール基を有するシルセスキオキサン化合物(a−1)とエチレン性不飽和結合を有する化合物およびイソシアネート基を有する化合物の少なくとも一方(a−2)とを混合し、硬化後の屈折率が1.53〜1.57である硬化性樹脂組成物(A)を得る工程と、
平均繊維径が45〜65nmであり、かつ繊維径分布が1〜1.3であるキチンナノファイバーを絡合させてキチンナノファイバー不織布(B)を得る工程と、
前記硬化性樹脂組成物(A)を前記キチンナノファイバー不織布(B)に含浸させてプリプレグを得る工程と、
前記プリプレグを硬化させる工程と、
を含む、キチンナノファイバー複合材料の製造方法。
【図1】

【公開番号】特開2013−112721(P2013−112721A)
【公開日】平成25年6月10日(2013.6.10)
【国際特許分類】
【出願番号】特願2011−258728(P2011−258728)
【出願日】平成23年11月28日(2011.11.28)
【出願人】(598045058)株式会社サムスン横浜研究所 (294)
【Fターム(参考)】
【公開日】平成25年6月10日(2013.6.10)
【国際特許分類】
【出願日】平成23年11月28日(2011.11.28)
【出願人】(598045058)株式会社サムスン横浜研究所 (294)
【Fターム(参考)】
[ Back to top ]