
新規α−置換プロピオン酸誘導体
【課題】新規α−置換プロピオン酸誘導体及び、該α−置換プロピオン酸誘導体PPARγに選択的なアゴニストとして機能するα−置換プロピオン酸誘導体。
【解決手段】下記一般式(I)で表されるα−置換プロピオン酸誘導体。

[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)等、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R5は置換基を有していても良いフェニル基又はシクロヘキシル基、nは0から10の整数を表す]
【解決手段】下記一般式(I)で表されるα−置換プロピオン酸誘導体。
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)等、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R5は置換基を有していても良いフェニル基又はシクロヘキシル基、nは0から10の整数を表す]
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規α−置換プロピオン酸誘導体、及び、該α−置換プロピオン酸誘導体であって、ペルオキシゾーム増殖薬活性化受容体(PPAR)γに選択的なアゴニストに関し、さらに、PPARγ活性の異常に起因する疾患等の治療及び/又は予防のための医薬又は医薬組成物、並びに、PPARγ活性の異常に起因する疾患等の治療及び/又は予防方法に関する。
【背景技術】
【0002】
ペルオキシソーム増殖剤応答性受容体(Peroxisome proliferator−activated receptor:PPAR、以下PPARとする)は、核内受容体スーパーファミリーに属するリガンド依存性の転写因子であり、標的遺伝子の転写をリガンド依存的に誘導する。すなわち、リガンドがPPARに結合すると、PPARは標的遺伝子のプロモーター領域に存在するPPAR応答配列(PPAR responsive element:PPRE)に結合し、標的遺伝子の転写が誘導される。
細胞内において、PPARはレチノイドXレセプター(RXR)とヘテロ二量体を形成する。このヘテロ二量体がPPAR応答配列(PPRE)として知られるDNA配列に結合して、各種遺伝子の転写を活性化する。また、PPAR/RXRヘテロ二量体は、DRIP−205やSRC−1などの活性化補助因子を取り込んで、標的遺伝子にコードされるmRNAの発現レベルを調節する。
【0003】
これまでに組織分布を異にする3種類のサブタイプ(α型、β/δ型、γ型)がヒトをはじめとする様々な動物種で同定されている。これらのうち、PPARαは、脂肪酸の異化能の高い肝臓、腎臓、心臓及び筋肉等に分布しており、特に肝臓において高発現が認められ、PPARαによって標的遺伝子の転写が誘導されると、血中中性脂肪の低下、HDLコレステロールの増加、体重の減少、血管新生の促進等が誘起される。PPARβ/δは、骨格筋を中心に生体内各組織に普遍的に発現しており、PPARβ/δの活性化により、骨格筋における脂肪酸の異化、HDLコレステロールの増加、インスリン抵抗性の改善、肥満の抑制などが誘導されることが明らかとなってきている(非特許文献1、非特許文献2)。
【0004】
PPARγは、脂肪細胞やマクロファージに高発現しており、脂肪細胞の分化、インスリン感受性の獲得などに関与するタンパク質を誘導する。アイソフォームとして、PPARγ1、γ2及びγ3の少なくとも3種類の存在が確認されており、これらは、選択的スプライシングの結果、発現すると考えられている。また、PPARγは、大腸癌、乳癌、前立腺癌、膵臓癌、肺癌、骨髄性白血病細胞及びリンパ性白血病細胞などのおおくの癌細胞においても、その発現が確認されている(非特許文献3〜5)。さらに、PPARγのリガンドであるチアゾリジンジオン(TZDs)が種々の異なるタイプの腫瘍細胞(胃腸、胆管及び膵臓の腺癌)の増殖を阻害することが報告されており(非特許文献4)、PPARγが抗癌剤のターゲットとして、にわかに注目をあびつつある。PPARγによる抗腫瘍機構については、種々の組織由来の腫瘍毎に異なることも予想され、その全容は未だ明らかにされていない。ただ、PPARγが、サイクリンD1の分解を促進しG1期で細胞増殖を停止させること(非特許文献6)、E2F−DPのDNA結合を阻害してその転写活性を抑制する(非特許文献7)などの報告から、PPARγが細胞増殖の抑制に直接関与していることが示唆されている。さらに、PPARγの活性が上昇すると血管新生因子であるVEGFの産生が減少することなども報告されており、PPARγの血管新生阻害作用も考慮されている(非特許文献8)。
【0005】
PPARの各サブタイプは、各々のターゲット遺伝子の転写を誘導することにより、脂肪代謝、インスリン抵抗の改善など、いわゆる、メタボリック症候群として知られる諸症状の緩和、あるいは、癌細胞の増殖抑制などに寄与していることが予想されている。すでに、PPARαに対する外因性リガンドとして、フェノフィブラート、ベザフィブラート、クロフィブラートなどのいわゆるフィブラート系の薬剤が、また、PPARγに対する外因性リガンドとして、トログリタゾン、ピオグリタゾン、ロシグリタゾンなどのいわゆるチアゾリジン系の薬剤が知られている。また、フィブラート系、チアゾリジン系以外で、PPARの各アイソフォームをターゲットとする化合物もいくつか報告されている(特許文献1〜6を参照のこと)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】WO01/092201号
【特許文献2】WO00/75103号
【特許文献3】WO2004/056748号
【特許文献4】WO2004/046091号
【特許文献5】WO03/051821号
【特許文献6】WO02/098840号
【非特許文献】
【0007】
【非特許文献1】Oliverら、Proc.Natl.Acad.Sci.USA. 98:5306−5311 2001
【非特許文献2】Tanakaら、Proc.Natl.Acad.Sci.USA. 100:15924−15929 2003
【非特許文献3】Smithら、Invest New Drugs.20:195−200 2002
【非特許文献4】Tsujieら、Exp.Cell Res.289:143−151 2003
【非特許文献5】Campbellら、PPAR Research Volume 2008,Article ID 314974
【非特許文献6】Qinら、Cancer Res.63 958−964 2003
【非特許文献7】Altiokら、Genes Dev.11 1987−1988 1997
【非特許文献8】Panigraphyら、J.Clin.Invest.110 923−932 2002
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は、新規α−置換プロピオン酸誘導体、及び該α−置換プロピオン酸誘導体であってPPARγに選択的なアゴニストの提供を目的とする。
また、本発明は、該α−置換プロピオン酸誘導体を有効成分として含むPPARγ転写活性化剤の提供を目的とする。
さらに、本発明は、該α−置換プロピオン酸誘導体を有効成分として含む、PPARγ転写活性の異常に起因する疾患(例えば、癌など)の治療剤及び/又は予防剤の提供を目的とする。
【課題を解決するための手段】
【0009】
上述の通り、脂肪代謝異常などのメタボリック症候群や癌の治療薬として注目を浴びているPPARアゴニストであるが、解決すべき問題も存在している。既存のPPARアゴニスト(例えば、PPARγアゴニスト)の中には、その強力なアゴニスト活性に基づく副作用(例えば、肝機能障害、浮腫、体重増加など)を誘発する化合物が報告されている(特開2007−314464明細書などを参照)。そのため、現段階においても、より薬理効果が高く、副作用(絶対的又は相対的に)等の少ないPPARアゴニスト及びこれを含む治療薬の開発が望まれている。
本発明者らは、上記事情に鑑み、鋭意研究を行ったところ、新規α−置換プロピオン酸誘導体を合成し、該α−置換プロピオン酸誘導体であってPPARγに選択的な高いアゴニスト活性を示す化合物を見出し、本発明を完成させた。
【0010】
すなわち本発明は、 一般式(I):
【化1】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、nは0から10の整数を表す]で表されるα−置換フェニルプロピオン酸誘導体若しくはその薬学上許容される塩又は水和物である。
さらに、本発明は上記一般式(I)で表されるα−置換フェニルプロピオン酸誘導体であるPPARγに選択的なアゴニストである。
【0011】
また、本発明は、上記一般式(I)で表されるα−置換フェニルプロピオン酸誘導体若しくはその薬学上許容される塩又は水和物を含有する、PPARγ転写活性化剤である。
【0012】
さらに、本発明は、上記一般式(I)で表されるα−置換フェニルプロピオン酸誘導体若しくはその薬学上許容される塩又は水和物を含有する医薬である。
【0013】
また、本発明は、上記一般式(I)で表される化合物の製造に適する、以下の一般式(II)及び一般式(III)の化合物である。
【化2】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基を表す]
【化3】
[式中、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R4は水素原子又はハロゲン原子、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、Pは、不斉補助基又は炭素数1から10の直鎖状又は分岐状アルキル基、nは0から10の整数を表す]
【発明の効果】
【0014】
本発明により、新規α−置換フェニルプロピオン酸誘導体及び該α−置換フェニルプロピオン酸誘導体であってPPARγに選択的なアゴニストが提供される。また、本発明により、PPARγ転写活性化剤が提供される。
【0015】
さらに、本発明により、PPARγ転写活性の異常によって惹起される疾患、例えば、癌、メタボリック症候群(例えば、糖尿病、高脂血症、高コレステロール症、高血圧症、肥満症、動脈硬化症など)などの予防又は治療剤が提供される。
【0016】
また、本発明の一般式(I)で示される化合物は、一般式(II)又は一般式(III)の中間体化合物を使用することで、迅速かつ効率的な製造が可能となる。
【図面の簡単な説明】
【0017】
【図1】本発明の化合物の癌細胞の増殖への効果。 本発明の化合物(実施例1の化合物)の癌細胞(OCUM−2MD3)に対するアポトーシス誘導効果(アポトーシス)及び増殖抑制効果(増殖抑制)を示す。実施例1の化合物は、0.1、1、10、100μM使用した。対照として、DMSOのみ(cont)、あるいは、トログリタゾン(Tro;10μM、100μM)を添加して実験を行った。
【図2】本発明の化合物の正常細胞への影響。 本発明の化合物(実施例1、実施例13及び実施例15の化合物)の癌細胞(OCUM−2MD3)及び正常細胞(OUMS−24)に対するアポトーシス誘導率を示す。
【発明を実施するための形態】
【0018】
本発明の実施形態の1つは、一般式(I)で表されるα−置換プロピオン酸誘導体若しくはその薬学上許容される塩又は水和物、あるいは、該α−置換プロピオン酸誘導体であってPPARγに選択的なアゴニストである。ここで、「アゴニスト」とは、PPARγに結合して、PPARγによる標的遺伝子の転写活性化を誘導する化合物のことである。
一般式(I)において、R1は、アダマンチル基、無置換又は置換基を有していても良いフェニル基、無置換又は置換基を有していても良いピリジル基、無置換又は置換基を有していても良いピリミジニル基、無置換又は置換基を有していても良いピラジニル基、無置換又は置換基を有していても良いピペリジン基、無置換又は置換基を有していても良いアゼピル基、無置換又は置換基を有していても良いピペラジニル基であり、あるいは、2−チエニル基、モルホリノ基である。ここで、置換基を有するフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基、ピペラジニル基の置換基としては、例えば、ハロゲン原子が好ましく、特に、フッ素原子が好ましい。
R2及びR4は、同一又は相異なって、水素原子又はハロゲン原子であり、好ましくは、水素原子又はフッ素原子である。
R3は炭素数1から10の直鎖状又は分岐状アルキル基、好ましくは、炭素数1〜5の直鎖状アルキル基、より好ましくは、プロピル基である。
R5は置換基を有していても良いフェニル基又はシクロヘキシル基であり、置換基を有するフェニル基又はシクロヘキシル基の置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基であり、好ましくは、フェニル基、フルオロフェニル基、シクロヘキシル基であり、より好ましくは、フェニル基又はフルオロフェニル基である。R5の置換基の位置は任意であるが、例えば、2位、4位などが好ましい。nは0〜10の整数を表し、好ましくは、1〜3の整数である。
【0019】
一般式(I)で表される本発明の化合物は、一般式(I)で表されるα−置換プロピオン酸誘導体のみならず、その塩又はそれらの溶媒和物若しくは水和物であっても良い。α−置換プロピオン酸誘導体の塩は特に限定されるものではなく、慣用の塩、例えば、ナトリウム塩、カリウム塩、リチウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アルミニウム塩等の金属塩が挙げられ、好ましくは薬学上許容されるものである。
また、一般式(I)で表されるα−置換プロピオン酸誘導体には、特に断らない限り、その互変異性体、幾何異性体(例えば、E体、Z体など)、鏡像異性体等の立体異性体も含まれる。すなわち、一般式(I)で表されるα−置換プロピオン酸誘導体中に、1個又は2個以上の不斉炭素が含まれる場合、不斉炭素の立体化学については、それぞれ独立して(R)体又は(S)体のいずれかをとることができ、該誘導体の鏡像異性体又はジアステレオ異性体などの立体異性体として存在することがある。
本発明の医薬の有効成分としては、純粋な形態の任意の立体異性体、立体異性体の任意の混合物、ラセミ体などを用いることが可能である。
【0020】
また、本発明の他の実施形態は、一般式(II)又は一般式(III)で示される化合物である。一般式(II)又は一般式(III)で示される化合物は、一般式(I)のα−置換プロピオン酸誘導体を製造するのに適した化合物である。
【0021】
一般式(II)において、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、無置換又は置換基を有していても良いピリジル基、無置換又は置換基を有していても良いピリミジニル基、無置換又は置換基を有していても良いピラジニル基、無置換又は置換基を有していても良いピペリジン基、無置換又は置換基を有していても良いアゼピル基、無置換又は置換基を有していても良いピペラジニル基であり、あるいは、2−チエニル基、モルホリノ基であり、置換基を有するフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基、ピペラジニル基の置換基としては、例えば、ハロゲン原子が好ましく、特に、フッ素原子が好ましい。好ましいR1として、アダマンチル基、ピリミジン−2−イル基、ピペリジニル基を挙げることができる。
R2は水素原子又はハロゲン原子であり、好ましくはハロゲン原子、特に好ましくはフッ素原子である。R3は炭素数1から10の直鎖状又は分岐状アルキル基、好ましくは、炭素数1〜5の直鎖状アルキル基、より好ましくは、プロピル基である。R4は水素原子又はハロゲン原子を表し、好ましくは水素原子又はフッ素原子である。
【0022】
一般式(III)において、R3は炭素数1から10の直鎖状又は分岐状アルキル基、好ましくは、炭素数1〜5の直鎖状アルキル基、より好ましくは、プロピル基である。R4は水素原子又はハロゲン原子を表し、好ましくは、水素原子又はフッ素原子である。R5は置換基を有していても良いフェニル基又はシクロヘキシル基であり、置換基を有するフェニル基又はシクロヘキシル基の置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基であり、好ましくは、フェニル基、フルオロフェニル基、シクロヘキシル基であり、より好ましくは、フェニル基又はフルオロフェニル基である。R5の置換基の位置は任意であるが、例えば、2位、4位などが好ましい。nは0〜10の整数を表し、好ましくは、1〜3の整数である。Pは、不斉補助基である。不斉補助基としては、反応を妨げない限り既知のものを適宜使用することができる。例えば、4−ベンジルオキサゾリジノン、4−イソプロピルオキサゾリジノン等を挙げることができる。
【0023】
一般式(I)で示されるPPARγに選択的なアゴニスト及びその塩としては、限定はしないが、例えば、次のものが挙げられる。
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸、
(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸、
(S)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸、
(R)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸、
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸、
(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸、
(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
(3−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
(4−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
2−ベンジル−3−(2−フルオロ−5−((2−フルオロ−4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸、
2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸。
(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(ピリミジン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸
(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(チオフェン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸
(R)−2−ベンジル−3−(3−((4−(ピペリジン−1−イル)ベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−((2’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−((3’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−((2’6’ジフルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−(2’−メチルビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−(ビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
【0024】
本発明の一般式(I)で表される化合物のうち、R4が水素原子、R5が無置換フェニル基である光学活性な化合物(Ia)は、以下の方法により製造することができる(スキーム1)。
【化4】
【0025】
すなわち、一般式(Ia):
【化5】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、nは0から10の整数を表す]
で表される化合物は、以下の:
【化6】
[式中、R3は前記と同義である]で表される化合物(A)と、以下の:
【化7】
[式中、nは前記と同義である]で表される化合物(G)とを反応させ(第一工程)、得られた以下の:
【化8】
[式中、R3及びnは前記と同義である]で表される化合物(B)を還元する(第二工程)ことにより製造することができる以下の:
【化9】
[式中、R3及びnは前記と同義である]で表される化合物(C)を還元する(第三工程)ことにより製造することができる以下の:
【化10】
[式中、R3及びnは前記と同義である]で表される化合物(D)を酸化する(第四工程)ことにより製造することができる以下の:
【化11】
[式中、R3及びnは前記と同義である]で表される化合物(E)と以下の:
【化12】
[式中、R1及びR2は前記と同義である]で表わされる化合物(H)を還元的アミドアルキル化(第五工程)ことにより製造することができる以下の:、
【化13】
[式中、R1、R2、R3およびnは前記と同義である]で表わされる化合物(F)の不斉補助基を脱保護する(第六工程)ことにより合成することができる。
【0026】
第一工程の反応は、テトラヒドロフランやジエチルエーテル、ヘキサン等の溶媒中塩基として例えば水素化ナトリウムのようなアルカリ金属水素化物、ブチルリチウムのような有機金属化合物、リチウムジイソプロピルアミド、ナトリウムビス(トリメチルシリル)アミドのような金属アミドを用いる事ができる。反応温度としては−100℃から室温にて、好適には−80℃から0℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0027】
第二工程の反応はパラジウム担持活性炭、白金担持活性炭、酸化白金、ロジウム担持アルミナ等の金属触媒存在下、エタノール、メタノール、テトラヒドロフラン、酢酸エチル、N,N−ジメチルホルムアミド等の溶媒中水素圧98.1kPaから491kPaで実施する事ができる。反応温度としては0℃から150℃にて、好適には室温から100℃にて実施する事ができる。反応時間は通常0.5〜24時間、好ましくは1〜5時間である。
【0028】
第三工程の反応は、テトラヒドロフランやジエチルエーテル、ヘキサン等の溶媒中塩基として例えばボランTHFコンプレックス、ジイソブチルアルミニウムヒドリド等の塩基を用いて実施する事ができる。反応温度としては−50℃から100℃にて、好適には−20℃から50℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0029】
第四工程の反応は、塩化メチレンやクロロホルム等の溶媒中酸化剤として例えば活性化二酸化マンガン、ピリジニウムクロロクロメート、ピリジニウムジクロメート等の塩基酸化剤を用いて実施する事ができる。反応温度としては−50℃から100℃にて、好適には−0℃から溶媒の還流温度にて実施する事ができる。反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0030】
第五工程の反応は、トルエン、ベンゼン、アセトニトリル、ジオキサン、N,N−ジメチルホルムアミド等の溶媒中、酸(例えば、トリフルオロ酢酸、トリフルオロメタンスルホン酸)の存在下、還元剤としてトリエチルシランの存在下で実施することができる。この際の反応温度は通常−20〜150℃、好ましくは0〜100℃であり、反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0031】
第六工程の反応は、アルカリ性条件下で実施する事ができる。アルカリ性条件としては水酸化リチウムと過酸化水素の混合物が用いられる。反応温度としては−20℃から100℃にて、好適には0℃から50℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0032】
また、本発明の一般式(I)で表される化合物のうち、R1がアダマンチル基、R2及びR4がフッ素原子、R3がプロピル基、R5が水素原子、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基を有するフェニル基又はシクロヘキシル基、nが1である化合物(Ib)は、以下の方法によって製造することができる(スキーム2)。
【化14】
【0033】
すなわち、一般式(Ib):
【化15】
[式中、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)を表す]で表される化合物は、以下の:
【化16】
で表される化合物(I)をホルミル化(第七工程)して得られる以下の:
【化17】
化合物(J)と以下の:
【化18】
化合物(O)とを還元的アミドアルキル化(第八工程)して得られる以下の:
【化19】
化合物(K)をホルミル化する(第九工程)ことにより製造することができる以下の:
【化20】
化合物(L)と以下の:
【化21】
[式中、R5は前記と同義である]で表される化合物(P)とを反応させる(第十工程)ことにより製造することができる以下の:
【化22】
[式中、R5は前記と同義である]で表される化合物(M)を還元する(第十一工程)ことにより製造することができる以下の:
【化23】
[式中、R5は前記と同義である]で表わされる化合物(N)を加水分解(第十二工程)することにより製造することができる。
【0034】
第七工程の反応は、ジクロロメタンやクロロホルム、四塩化炭素等の溶媒中ホルミル化剤としてジクロロメチルメチルエーテルを用い、さらにルイス酸として四塩化チタンやチタニウムテトライソプロポキシド、塩化アルミニウムを用いて製造する事ができる。反応温度としては−100℃から室温にて、好適には−80℃から0℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0035】
第八工程の反応は、トルエン、ベンゼン、アセトニトリル、ジオキサン、N,N−ジメチルホルムアミド等の溶媒中、酸(例えば、トリフルオロ酢酸、トリフルオロメタンスルホン酸)の存在下、還元剤としてトリエチルシランの存在下で実施することができる。この際の反応温度は通常−20〜150℃、好ましくは0〜100℃であり、反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0036】
第九工程の反応は、ジクロロメタンやクロロホルム、四塩化炭素等の溶媒中ホルミル化剤としてジクロロメチルメチルエーテルを用い、さらにルイス酸として四塩化チタンやチタニウムテトライソプロポキシド、塩化アルミニウムを用いて製造する事ができる。反応温度としては−100℃から室温にて、好適には−80℃から0℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0037】
第十工程の反応はテトラヒドロフラン、トルエン、ジオキサン、N,N−ジメチルホルムアミド等の溶媒中、塩基としては例えば水素化ナトリウムのようなアルカリ金属水素化物、ブチルリチウムのような有機金属化合物、リチウムジイソプロピルアミドのような金属アミド、ナトリウムメトキシドやカリウム t−ブトキシドのような金属アルコキシドを用いる事ができる。反応温度としては−20℃から150℃にて、好適には0℃から50℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0038】
第十一工程の反応は、パラジウム担持活性炭、白金担持活性炭、酸化白金、ロジウム担持アルミナ等の金属触媒存在下、エタノール、メタノール、テトラヒドロフラン、酢酸エチル、N,N−ジメチルホルムアミド等の溶媒中水素圧1kgf/cm2から5kgf/cm2で実施する事ができる。反応温度としては0℃から100℃にて、好適には室温から80℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0039】
第十二工程の反応は、アルカリ性条件下で行う事ができる。アルカリ性条件としては水酸化リチウム、水酸化ナトリウム、水酸化カリウム等が用いられる。反応温度としては0℃から80℃にて、好適には室温から60℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0040】
本発明には、一般式(I)で示されるα−置換プロピオン酸誘導体を含むPPARγ転写活性化剤(PPARγ標的遺伝子の転写を活性化(又は誘導)する薬剤)が含まれる。
【0041】
さらに、本発明のα−置換プロピオン酸誘導体であってPPARγに選択的なアゴニストを含む医薬も本発明の範囲に含まれる。
本発明の医薬は、PPARγの異常調節、特に、その活性が抑制されることによって発症する疾患等の治療剤又は予防剤として使用することができる。PPARγの既存のアゴニストであるチアゾリジン誘導体がインスリン抵抗性を改善し(例えば、Yamauchiら, J. Biol. Chem., 276:41245-41254, 2001など)、血糖値の調製に有効であることの他、血中遊離脂肪酸、TGの低下作用、HDL−C増加作用などが確認されており(例えば、Leeら, Endocrinology, 144:2201-2207, 2003など)、PPARγのアゴニストは糖尿病、高脂血症など、メタボリックシンドロームに関連する疾患の治療等に利用できることが知られている。さらに、PPARγは、大腸癌、乳癌、前立腺癌、膵臓癌、肺癌、骨髄性白血病細胞及びリンパ性白血病細胞などの多くの癌細胞においても、その発現が確認されている(非特許文献3〜5)。また、PPARγのアゴニストであるチアゾリジンジオン(TZDs)が種々の異なるタイプの腫瘍細胞(胃腸、胆管及び膵臓の腺癌)の増殖を阻害することなどから(非特許文献4)、PPARγのアゴニストは、抗癌剤としても使用することができる。
【0042】
一般式(I)で示されるPPARγに選択的なアゴニストを含む医薬は、脂質代謝異常、高脂血症、糖尿病、高血圧、高コレステロール症、肥満症、などのメタボリックシンドローム、並びに、メタボリックシンドロームを基盤として発症する、例えば、動脈硬化性疾患などの治療剤又は予防剤として使用することができる。さらに、一般式(I)で示されるPPARγに選択的なアゴニストを含む医薬は、癌の治療剤又は予防剤、例えば、大腸癌、乳癌、前立腺癌、膵臓癌、肺癌、骨髄性白血病細胞及びリンパ性白血病細胞の他、特に、胃腸、胆管及び膵臓などに発症する腺癌の治療剤又は予防剤として使用することができる。また、一般式(I)で示される化合物を含む本発明の医薬は、胃腺癌のうち印環細胞癌(進行型の低分化腺癌、非充実型(スキルス;Scirrhous)などを含む)などの治療にも使用することができる。
【0043】
本発明の医薬の有効成分としては、上記一般式(I)で表される化合物のほか、生理学的に許容されるその塩を用いても良い。塩としては、例えば、酸性基が存在する場合には、リチウム、ナトリウム、カリウム、マグネシウム、カルシウム等のアルカリ金属及びアルカリ土類金属塩;アンモニア、メチルアミン、ジメチルアミン、トリメチルアミン、ジシクロヘキシルアミン、トリス(ヒドロキシメチル)アミノメタン、N,N−ビス(ヒドロキシエチル)ピペラジン、2−アミノ−2−メチル−1−プロパノール、エタノールアミン、N−メチルグルカミン、L−グルカミン等のアミンの塩;又はリジン、δ−ヒドロキシリジン、アルギニンなどの塩基性アミノ酸との塩を形成することができる。塩基性基が存在する場合には、塩酸、臭化水素酸、硫酸、硝酸、リン酸等の鉱酸の塩;メタンスルホン酸、ベンゼンスルホン酸、パラトルエンスルホン酸、酢酸、プロピオン酸塩、酒石酸、フマル酸、マレイン酸、リンゴ酸、シュウ酸、コハク酸、クエン酸、安息香酸、マンデル酸、ケイ皮酸、乳酸、グリコール酸、グルクロン酸、アスコルビン酸、ニコチン酸、サリチル酸等の有機酸との塩;又はアスパラギン酸、グルタミン酸などの酸性アミノ酸との塩などを挙げることができる。
さらに、本発明の医薬の有効成分として、一般式(I)で表される化合物又はその塩の溶媒和物若しくは水和物を用いることもできる。
【0044】
本発明の医薬は、有効成分である一般式(I)で表される化合物及び薬理学的に許容されるその塩、又はそれらの溶媒和物若しくはそれらの水和物自体を投与しても良いが、一般的には、有効成分である一般式(I)の化合物と1又は2以上の製剤用添加物とを含む医薬組成物の形態で投与することが望ましい。本発明の医薬の有効成分としては、一般式(I)の化合物の2種以上を組み合わせて用いることができ、上記医薬組成物には、癌、糖尿病、高脂血症、高コレステロール症、高血圧症、肥満症、動脈硬化症などの諸症状の予防又は治療のための他の既知の有効成分を配合することも可能である。
【0045】
医薬組成物の種類は特に限定されず、剤型としては、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤、懸濁剤、座剤、軟膏、クリーム剤、ゲル剤、貼付剤、吸入剤、注射剤等が挙げられる。これらの製剤は常法に従って調製される。なお、液体製剤にあっては、用時、水又は他の適当な溶媒に溶解又は懸濁する形であっても良い。また錠剤、顆粒剤は周知の方法でコーティングしても良い。注射剤の場合には、本発明の化合物を水に溶解させて調製されるが、必要に応じて生理食塩水あるいはブドウ糖溶液に溶解させてもよく、また緩衝剤や保存剤を添加しても良い。経口投与用又は非経口投与用の任意の製剤形態で提供される。例えば、顆粒剤、細粒剤、散剤、硬カプセル剤、軟カプセル剤、シロップ剤、乳剤、懸濁剤又は液剤等の形態の経口投与用医薬組成物、静脈内投与用、筋肉内投与用、若しくは皮下投与用などの注射剤、点滴剤、経皮吸収剤、経粘膜吸収剤、点鼻剤、吸入剤、坐剤などの形態の非経口投与用医薬組成物として調製することができる。注射剤や点滴剤などは、凍結乾燥形態などの粉末状の剤形として調製し、用時に生理食塩水などの適宜の水性媒体に溶解して用いることもできる。また、高分子などで被覆した徐放製剤を脳内に直接投与することも可能である。
【0046】
医薬組成物の製造に用いられる製剤用添加物の種類、有効成分に対する製剤用添加物の割合、又は医薬組成物の製造方法は、組成物の形態に応じて当業者が適宜選択することが可能である。製剤用添加物としては無機又は有機物質あるいは固体又は液体の物質を用いることができ、一般的には、有効成分重量に対して1重量%から90重量%の間で配合することができる。具体的には、その様な物質の例として乳糖、ブドウ糖、マンニット、デキストリン、シクロデキストリン、デンプン、蔗糖、メタケイ酸アルミン酸マグネシウム、合成ケイ酸アルミニウム、カルボキシメチルセルロースナトリウム、ヒドロキシプロピルデンプン、カルボキシメチルセルロースカルシウム、イオン交換樹脂、メチルセルロース、ゼラチン、アラビアゴム、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、ポリビニルアルコール、軽質無水ケイ酸、ステアリン酸マグネシウム、タルク、トラガント、ベントナイト、ビーガム、酸化チタン、ソルビタン脂肪酸エステル、ラウリル硫酸ナトリウム、グリセリン、脂肪酸グリセリンエステル、精製ラノリン、グリセロゼラチン、ポリソルベート、マクロゴール、植物油、ロウ、流動パラフィン、白色ワセリン、フルオロカーボン、非イオン性界面活性剤、プロピレングルコール、水等が挙げられる。
【0047】
経口投与用の固形製剤を製造するには、有効成分と賦形剤成分例えば乳糖、澱粉、結晶セルロース、乳酸カルシウム、無水ケイ酸などと混合して散剤とするか、さらに必要に応じて白糖、ヒドロキシプロピルセルロース、ポリビニルピロリドンなどの結合剤、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウムなどの崩壊剤などを加えて湿式又は乾式造粒して顆粒剤とする。錠剤を製造するには、これらの散剤及び顆粒剤をそのまま或いはステアリン酸マグネシウム、タルクなどの滑沢剤を加えて打錠すれば良い。これらの顆粒又は錠剤はヒドロキシプロピルメチルセルロースフタレート、メタクリル酸−メタクリル酸メチルポリマーなどの腸溶剤基剤で被覆して腸溶剤製剤あるいはエチルセルロース、カルナウバロウ、硬化油などで被覆して持続性製剤とすることもできる。また、カプセル剤を製造するには、散剤又は顆粒剤を硬カプセルに充填するか、有効成分をそのまま或いはグリセリン、ポリエチレングリコール、ゴマ油、オリーブ油などに溶解した後ゼラチン膜で被覆し軟カプセルとすることができる。
【0048】
注射剤を製造するには、有効成分を必要に応じて塩酸、水酸化ナトリウム、乳糖、乳酸、ナトリウム、リン酸一水素ナトリウム、リン酸二水素ナトリウムなどのpH調整剤、塩化ナトリウム、ブドウ糖などの等張化剤と共に注射用蒸留水に溶解し、無菌濾過してアンプルに充填するか、更にマンニトール、デキストリン、シクロデキストリン、ゼラチンなどを加えて真空凍結乾燥し、用事溶解型の注射剤としても良い。また、有効成分にレチシン、ポリソルベート80、ポリオキシエチレン硬化ヒマシ油などを加えて水中で乳化せしめ注射剤用乳剤とすることもできる。
【0049】
直腸投与剤を製造するには、有効成分をカカオ脂、脂肪酸のトリ、ジ及びモノグリセリド、ポリエチレングリコールなどの座剤用基材と共に加湿して溶解し型に流し込んで冷却するか、有効成分をポリエチレングリコール、大豆油などに溶解した後、ゼラチン膜で被覆すれば良い。
【0050】
本発明の医薬の投与量及び投与回数は特に限定されず、治療対象疾患の悪化・進展の防止及び/又は治療の目的、疾患の種類、患者の体重や年齢、疾患の重篤度などの条件に応じて、医師の判断により適宜選択することが可能である。一般的には、経口投与における成人一日あたりの投与量は0.01〜1000mg(有効成分重量)程度であり、一日1回又は数回に分けて、或いは数日ごとに投与することができる。注射剤として用いる場合には、成人に対して一日量0.001〜100mg(有効成分重量)を連続投与又は間欠投与することが望ましい。
【0051】
本発明の医薬は、植込錠及びマイクロカプセルに封入された送達システムなどの徐放性製剤として、体内から即時に除去されることを防ぎ得る担体を用いて調製することができる。担体として、例えば、エチレンビニル酢酸塩、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、及びポリ乳酸などの、生物分解性、生物適合性ポリマーを用いることができる。このような材料は、当業者によって容易に調製することができる。また、リポソームの懸濁液も薬剤的に受容可能な担体として使用することができる。有用なリポソームは、限定はしないが、ホスファチジルコリン、コレステロール及びPEG誘導ホスファチジルエタノール(PEG−PE)を含む脂質組成物として、使用に適するサイズになるように、適当なポアサイズのフィルターを通して調製され、逆相蒸発法によって精製される。
【0052】
本発明の医薬は、医薬組成物としてキットの形態で、容器、パック中に投与の説明書と共に含めることができる。本発明の医薬組成物がキットとして供給される場合、該組成物のうち異なる構成成分が別々の容器中に包装され、使用直前に混合される。このように構成成分を別々に包装するのは、活性構成成分の機能を失うことなく長期間の貯蔵を可能にするためである。
キット中に含まれる試薬は、構成成分の活性を長期間有効に持続し、容器内側に吸着することなく、また、構成成分を変質することのない材質で製造された容器中に供給される。例えば、封着されたガラスアンプルは、窒素ガスのような中性で不反応性を示すガスの存在下で封入されたバッファーなどを含んでも良い。アンプルは、ガラス、ポリカーボネート、ポリスチレンなどの有機ポリマー、セラミック、金属、又は試薬を保持するために通常用いられる他の何れかの適切な材料によって構成される。
【0053】
また、キットには使用説明書が添付されても良い。本キットの使用説明は、紙などに印刷され、及び/又はフロッピー(登録商標)ディスク、CD−ROM、DVD−ROM、Zipディスク、ビデオテープ、オーディオテープなどの電気的又は電磁的に読み取り可能な媒体に保存されて使用者に供給されても良い。詳細な使用説明は、キット内に実際に添付されていてもよく、あるいは、キットの製造者又は分配者によって指定され又は電子メール等で通知されるウェブサイトに掲載されていても良い。
【0054】
さらに、本発明には、PPARγ転写活性の異常に起因して発症する疾患等(例えば、癌の他、糖尿病、高脂血症、高コレステロール症、高血圧症、肥満症、動脈硬化症など)の予防又は治療方法が含まれる。
ここで「治療」とは、PPARγ転写活性の異常に起因して発症する疾患等に罹患した哺乳動物において、その病態の進行及び悪化を阻止又は緩和することを意味し、これによって該疾患の進行及び悪化を阻止又は緩和することを目的とする処置のことである。
また、「予防」とは、PPARγ転写活性の異常に起因して発症する疾患等に罹患するおそれがある哺乳動物について、該疾患の発症又は罹患を予め阻止することを意味し、これによって該疾患の諸症状等の発症を予め阻止することを目的とする処置のことである。
【0055】
治療の対象となる「哺乳動物」は、哺乳類に分類される任意の動物を意味し、特に限定はしないが、例えば、ヒトの他、イヌ、ネコ、ウサギなどのペット動物、ウシ、ブタ、ヒツジ、ウマなどの家畜動物などのことである。特に好ましい「哺乳動物」は、ヒトである。
【0056】
以下に実施例を示してさらに詳細に説明するが、本発明は実施例により何ら限定されるものではない。
【実施例】
【0057】
〔実施例1〕(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸〔化24〕の合成例
【化24】
式(I)の化合物の1つである(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸を以下の通り合成した。
【0058】
ベンジル 5−((R)−2−ベンジル−3−((S)−4−ベンジル−2−オキソオキサゾリジン−3−イル)−3−オキソプロピル)−2−プロポキシベンゾアートの合成
【化25】
(4S)−4−ベンジル−3−(3−フェニルプロパノイル)オキサゾリジン−2−オン(0.74g,2.37mmol)と脱水THF(20ml)をアルゴン雰囲気下混合した。反応液を−50℃に冷却し、LiHMDSのTHF溶液(8.0ml,8.0mmol)をシリンジで注加した。同温度で20分撹拌後、−15℃に昇温後さらに25分撹拌した。再度−50℃に冷却し、ベンジル 5−(ブロモメチル)−2−プロポキシ−ベンゾアート(0.86g,2.37mmol)を脱水THF(10ml)に溶かした溶液をゆっくり滴下した。反応液は75分かけて段階的に−50℃から0℃に昇温させた。0℃ にてさらに1時間撹拌後10%塩化アンモニウム水溶液中に反応液を注加し、酢酸エチル抽出(5×30ml)した。 抽出液は飽和食塩水で洗浄後無水硫酸マグネシウム乾燥し、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 9:2 v/v)、黄色油状の目的化合物を0.89g、収率64%で得た。
MS(FAB,(M+H)+) m/z 592 C37H37NO6
【0059】
5−((R)−2−ベンジル−3−((S)−4−ベンジル−2−オキソオキサゾリジン−3−イル)−3−オキソプロピル)−2−プロポキシ安息香酸の合成
【化26】
10% Pd/C (0.20g)、上記ベンジル 5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソプロピル]−2−プロポキシベンゾアート(0.89g,1.50mmol)及びAcOEt(80ml)を混合した。反応液を室温にて2.25時間接触還元を行った。反応液をセライトろ過し、ろ液を濃縮し、酢酸エチルで洗浄後ろ液と洗浄液を合わせ濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 5:2 v/v)、無色油状の目的化合物を0.41g、収率55%で得た。
1H NMR(400MHz,CDCl3)δ10.98(s,1H),8.06(d,J=2.4Hz,1H),7.52(dd,J=8.4,2.4Hz,1H),7.24−7.22(m,5H),7.20−7.16(m,3H),7.03(dd,J=7.2,2.0Hz,2H),6.97(d,J=8.4Hz,1H),4.56−4.48(m,1H),4.46−4.40(m,1H),4.22−4.13(m,2H),3.97(dd,J=8.8,2.4Hz,2H),3.81(t,J=8.2Hz,1H),3.16(dd,J=13.8,8.2Hz,1H),3.08−2.98(m,2H),2.84−2.76(m,2H),2.54(dd,J=13.4,9.4Hz,1H),1.96−1.88(m,2H),1.08(t,J=7.2Hz,3H);
MS(FAB,(M+H)+) m/z 502 C30H31NO6.
【0060】
(S)−4−ベンジル−3−((R)−2−ベンジル−3−(3−(ヒドロキシメチル)−4−プロポキシフェニル)プロパノイル)オキサゾリジン−2−オンの合成
【化27】
上記5−[(R)−2−ベンジルl−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシ安息香酸(0.41g,0.82mmol)と脱水THF(30ml)を混合した。アルゴン雰囲気下反応液を0℃に冷却し、1mol/LのBH3−THFコンプレックス1.60mL(1.60mmol)をシリンジで滴下した。反応液は0℃で2時間撹拌後室温にて一晩撹拌した。反応液を飽和塩化アンモニウム水溶液中に注加し、酢酸エチルにて抽出した。抽出液は飽和食塩水洗浄後無水硫酸マグネシウム乾燥し濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 3:1 v/v)、無色油状の目的化合物を0.40g、収率100%で得た。
1H NMR(400MHz,CDCl3)δ7.28−7.15(m,10H),6.99(dd,J=7.2,2.0Hz,2H),6.78(d,J=8.4Hz,1H),4.66(d,J=6.4Hz,2H),4.60−4.53(m,1H),4.43−4.38(m,1H),3.98−3.89(m,2H),3.77(t,J=8.2Hz,1H),3.08(dd,J=13.4,8.6Hz,1H),3.01−2.94(m,2H),2.85−2.75(m.2H),2.49(dd,J=13.4,9.4Hz,1H),2.37(t,J=6.6Hz,1H),1.85−1.76(m,2H),1.03(t,J=7.2Hz,3H);
MS(FAB,(M+H)+) m/z 488 C30H33NO5.
【0061】
5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシベンズアルデヒドの合成
【化28】
活性化二酸化マンガン(5.7g,65.6mmol)、上記(S)−4−ベンジル−3−[(R)−2−ベンジル−3−[3−(ヒドロキシメチル)−4−プロポキシ−フェニル]プロパノイル]オキサゾリジン−2−オン(0.40g,3.0mmol)とCH2Cl2(40ml)を混合し23時間還流した。反応液をアセトンで希釈しセライトを通して反応液を濾過し、ろ液を濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 3:1 v/v)、無色油状の目的化合物を0.20g、収率50%で得た。
1H NMR(400MHz,CDCl3)δ10.47(d,J=2.8Hz,1H),7.70(d,J=2.0Hz,1H),7.49(dd,J=8.4,2.4Hz,1H),7.27−7.16(m,8H),7.01(dd,J=7.4,1.8Hz,2H),6.90(d,J=8.8Hz,1H),4.56−4.48(m,1H),4.45−4.39(m,1H),4.03−3.97(m,2H),3.95(dd,J=8.8,2.4Hz,1H),3.80(t,J=8.4Hz,1H),3.12(dd,J=13.6,8.8Hz,1H),3.05−2.97(m,2H),2.79(dd,J=14.0,6.4Hz,2H),2.51(dd,J=13.2,9.2Hz,1H),1.89−1.80(m,2H),1.05(t,J=7.4Hz,3H);
MS(FAB,(M+H)+) m/z 486 C30H31NO5.
【0062】
4−(1−アダマンチル)−N−[[5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシフェニル]メチル]ベンズアミドの合成
【化29】
4−(1−アダマンチル)ベンズアミド(0.27g,1.03mmol)、上記5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシ−ベンズアルデヒド(0.20g,0.41mmol)、トリエチルシラン(0.16ml,1.03mmol)、TFA(0.08ml,1.03mmol)および脱水トルエン(30ml)を混合し、アルゴン雰囲気下49時間加熱還流した。水(100ml)に反応液を注加し、酢酸エチルで抽出した。抽出液は飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、ろ過後濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 2:1 v/v)、褐色油状の目的化合物を0.29g、収率97%で得た。
1H NMR(300MHz,CDCl3)δ7.61(d,J=8.7Hz,2H),7.35(d,J=8.7Hz,2H),7.28−7.16(m,10H),6.88−6.85(m,2H),6.80(d,J=8.4Hz,1H),6.71(t,J=5.7Hz,1H),4.70−4.52(m,3H),4.42−4.34(m,1H),3.98−3.88(m,2H),3.74(t,J=8.4Hz,1H),3.07(dd,J=13.8,8.7Hz,1H),3.01−2.84(m,2H),2.79(dd,J=13.8,6.3Hz,2H),2.48(dd,J=13.2,9.0Hz,1H),2.10(s,3H),1.89−1.72(m,14H),1.03(t,J=7.5Hz,3H);
MS(FAB,(M+H)+) m/z 725 C47H52N2O5.
【0063】
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸(実施例1の目的化合物)の最終合成過程
30 % H2O2(0.4 mL,4.0mmol)、上記合成の4−(1−アダマンチル)−N−[[5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシフェニル]メチル]ベンズアミド(290mg,0.4mmol)、THF(24ml)、水(6ml)を混合し、次いで、リチウムヒドロキシド モノヒドラート(100mg,2.38mmol)を水(2ml)に溶かし加えた。アルゴン雰囲気下、反応液を0℃にて、2.5時間、室温で3時間撹拌後NaHSO3(1.0g)をH2O(6ml)に溶かし反応液に加えた。反応液を10%塩酸で希釈し酢酸エチルで抽出した。抽出液は飽和食塩水で洗浄し、無水硫酸マグネシウム乾燥後、ろ過、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 2:1 v/v)、無色粉末状の目的化合物(実施例1の目的化合物)を0.19g、収率86%で得た。
mp 177−178℃.
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.1Hz,2H),7.39(d,J=8.4Hz,2H),7.26−7.14(m,6H),7.03(dd,J=8.1,1.8Hz,1H),6.77−6.70(m,2H),4.64−4.51(m,2H),3.94(t,J=6.5Hz,2H),3.01−2.71(m,5H),2.10(s,3H),1.90−1.72(m,14H),1.06(t,J=7.4Hz,3H);
MS(FAB,(M+H)+)m/z 566 C37H43NO4 .
[α]D −3°(c0.10,CH3CN,26℃).
【0064】
〔実施例2〕(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸〔化30〕の合成例
【化30】
実施例1と同様の操作により、実施例2の化合物を合成した。
mp 177−178℃.
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.1Hz,2H),7.39(d,J=8.1Hz,2H),7.26−7.14(m,6H),7.03(dd,J=8.1,1.8Hz,1H),6.77−6.70(m,2H),4.64−4.51(m,2H),3.94(t,J=6.5Hz,2H),3.01−2.71(m,5H),2.10(s,3H),1.90−1.72(m,14H),1.06(t,J=7.4Hz,3H);
MS(FAB,(M+H)+)m/z 566 C37H43NO4.
[α]D +3°(c0.10,CH3CN,26℃).
【0065】
〔実施例3〕(S)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸〔化31〕の合成例
【化31】
実施例1と同様の操作により、実施例3の化合物を合成した。
mp 140−141℃.
1H NMR(300MHz,CDCl3)δ7.69(d,J=8.4Hz,2H),7.39(d,J=8.1Hz,2H),7.26−7.13(m,6H),7.02(dd,J=8.4,2.1Hz,1H),6.76−6.70(m,2H),4.59(d,J=4.5Hz,2H),3.93(t,J=6.45Hz,2H),2.91(dd,J=13.1,7.7Hz,1H),2.77−2.53(m,4H),2.10(s,3H),2.04−1.72(m,17H),1.05(t,J=7.4Hz,3H);
MS(FAB,(M+H)+) m/z 580 C38H45NO4.
[α]D −4°(c0.25,CH3CN,22℃).
【0066】
〔実施例4〕(R)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸の〔化32〕合成例
【化32】
実施例1と同様の操作により、実施例4の化合物を合成した。
mp 140−141℃.
1H NMR(300MHz,CDCl3)δ7.69(d,J=8.4Hz,2H),7.39(d,J=8.1Hz,2H),7.26−7.13(m,6H),7.02(dd,J=8.4,2.1Hz,1H),6.76−6.69(m,2H),4.59(d,J=4.5Hz,2H),3.93(t,J=6.5Hz,2H),2.91(dd,J=13.1,7.7Hz,1H),2.77−2.53(m,4H),2.10(s,3H),2.04−1.72(m,17H),1.05(t,J=7.4Hz,3H);
MS(FAB,(M+H)+) m/z 580 C38H45NO4.
[α]D+4°(c0.25,CHCl3,22℃).
【0067】
〔実施例5〕(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸〔化33〕の合成例
【化33】
実施例1と同様の操作により、実施例5の化合物を合成した。
mp 177−178℃.
1H NMR(300MHz,CDCl3)δ7.70(d,J=8.4Hz,2H),7.39(d,J=8.4Hz,2H),7.14(s,1H),7.04(dd,J=8.4,2.1Hz,1H),6.78−6.70(m,2H),4.65−4.53(m,2H),3.95(t,J=6.5Hz,2H),2.87−2.64(m,3H),2.10(s,3H),1.90−1.54(m,20H),1.34−1.12(m,5H),1.06(t,J=7.4Hz,3H),0.92−0.73(m,2H);
MS(FAB,(M+H)+) m/z 572 C37H49NO4.
[α]D −5.5°(c0.50,CHCl3,23℃).
【0068】
〔実施例6〕(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸〔化34〕の合成例
【化34】
実施例1と同様の操作により、実施例6の化合物を合成した。
mp 177−178℃.
1H NMR(300MHz,CDCl3)δ7.70(d,J=8.4Hz,2H),7.39(d,J=8.4Hz,2H),7.14(s,1H),7.03(dd,J=8.4,2.4Hz,1H),6.78−6.70(m,2H),4.65−4.53(m,2H),3.95(t,J=6.5Hz,2H),2.87−2.64(m,3H),2.10(s,3H),1.90−1.54(m,20H),1.34−1.12(m,5H),1.06(t,J=7.4Hz,3H),0.92−0.73(m,2H);
MS(FAB,(M+H)+) m/z 572 C37H49NO4.
[α]D +5.5°(c0.50,CHCl3,21℃).
【0069】
〔実施例7〕(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸〔化35〕の合成例
【化35】
式(I)の化合物の1つである(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸〔化35〕の合成例を以下に示す。
式(I)を合成するための中間化合物である式(II)の化合物の1つである4−アダマンチル−2−フルオロ−N−(4−フルオロ−5−ホルミル−2−プロポキシベンジル)ベンズアミド〔化36〕を以下のように合成した。
【化36】
【0070】
3−フルオロ−1−プロポキシベンゼンの合成
【化37】
3−フルオロフェノール(2.00g,17.8mmol)とDMF(6ml)を混合し、1−ヨードプロパン(4.25g,25.0mmol)および無水炭酸カリウム、K2CO3(2.76g,20.0mmol)を加え、室温にて一晩撹拌した。反応液は飽和塩化アンモニウム水溶液中に注加し、塩化メチレンで抽出した。抽出液は飽和食塩水で洗浄し、無水硫酸マグネシウム乾燥後、ろ過、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン)、無色油状の目的化合物を2.49g、収率91%で得た。
1H−NMR(500MHz,CDCl3)δ7.20(q,J=7.7Hz,1H),6.69−6.59(m,3H),3.90(t,J=6.7Hz,2H),1.81(m, 2H),1.04(t,J=7.6Hz,3H)
【0071】
4−フルオロ−2−プロポキシベンズアルデヒドの合成
【化38】
上記3−フルオロ−1−プロポキシベンゼン(2.39g,15.5mmol)とCH2Cl2(100mL)を混合し−78℃に冷却した。次にTiCl4(5mL)、CHCl2OCH3(2mL)を順次加え同温度で30分撹拌した。室温に戻しさらに一晩撹拌した。反応液は2mol/L塩酸中に注加し、塩化メチレンにて抽出した。抽出液は飽和食塩水で洗浄し、無水硫酸マグネシウム乾燥後、ろ過、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 5:1 v/v)、無色油状の目的化合物を1.74g、収率62%で得た。
1H−NMR(500MHz,CDCl3)δ10.39(s,1H),7.83(t,J=7.9Hz,1H),6.70−6.64(m,2H),4.00(t,J=6.4Hz,2H),1.87(m,2H),1.06(t,J=7.6Hz,3H)
【0072】
4−アダマンチル−2−フルオロ−N−(4−フルオロ−2−プロポキシベンジル)ベンズアミドの合成
【化39】
上記4−フルオロ−2−プロポキシベンズアルデヒド(661mg,3.63mmol)、2−フルオロ−4−アダマンチルベンズアミド(〔化18〕)(1.02g,3.73mmol)、トルエン(0.5mL)を混合し、トリフルオロ酢酸(0.5mL)、トリエチルシラン(1.6mL)を順次加えた。アルゴン雰囲気下、80℃にて3日撹拌した。反応液を濃縮し、残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 10/1から5/1 v/v)、無色油状の目的化合物を1.05g、収率66%で得た。
1H−NMR(500MHz,CDCl3)δ8.04(t,J=8.5Hz,1H),7.30−7.23(m,3H),7.05(d,J=14.6Hz,1H),6.61−6.58(m,2H),4.61(d,J=5.5Hz,2H),3.95(t,J=6.4 Hz,2H),2.10(s,3H),1.90−1.72(m,14H),1.08(t,J=7.3Hz,3H);
MS (FAB) 440 (M+H)+
【0073】
次に、上記4−アダマンチル−2−フルオロ−N−(4−フルオロ−2−プロポキシベンジル)ベンズアミド(1.01g,2.30mmol)と、CH2Cl2(20mL)を混合し−78℃に冷却した。次にTiCl4(1.0mL)、CHCl2OCH3(0.3mL)を順次加え同温度で30分撹拌した。室温に戻しさらに3時間撹拌した。反応液は2mol/L塩酸中に注加し、塩化メチレンにて抽出した。抽出液は飽和食塩水で洗浄し、無水硫酸マグネシウム乾燥後、ろ過、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 5:1 v/v)、無色粉末の目的化合物(〔化36〕の化合物)を1.02g、収率62%で得た。
1H−NMR(500MHz,CDCl3)δ10.08(s,1H),8.04(t,J=8.5Hz,1H),7.83(d,J=7.9Hz,1H),7.26−7.21(m,2H),7.07(dd,J=14.6,1.8Hz,1H),6.62(d,J=12.2Hz,1H),4.64(d,J=5.5Hz,2H),4.04(t,J=6.7Hz,2H),2.11(s,3H),1.93−1.73(m,14H),1.09(t,J=7.3Hz,3H);
MS (FAB) 468 (M+H)+
【0074】
〔化36〕の合成と同様にして、式(II)の化合物の1つである4−アダマンチル−N−(5−ホルミル−2−プロポキシベンジル)ベンズアミド(〔化40〕の化合物)の合成も行った。
【化40】
1H NMR(300MHz,CDCl3)δ9.87(s,1H),7.86(d,J=2.1Hz,1H),7.82(dd,J=8.4,2.1Hz,1H),7.73(d,J=8.4Hz,2H),7.43(d,J=8.1Hz,1H),6.99(d,J=8.4Hz,1H),6.59(t,J=5.7Hz,1H),4.70(d,J=6.0Hz,2H),4.09(t,J=6.5Hz,2H),2.11(s,3H),1.90−1.73(m,14H),1.10(t,J=7.4Hz,3H)
MS(FAB,(M+H)+) m/z 432 C28H33NO3
【0075】
メチル 2−(ジメトキシホスホリル)−3−(2−フルオロフェニル)プロパノアートの合成
【化41】
カリウムtert−ブトキシド(0.36g,3.2mmol)の脱水THF(20mL)溶液に0℃、アルゴン雰囲気下、ホスホノ酢酸トリメチル(0.43mL,2.7 mmol)を加え30分撹拌した。次に2−フルオロベンジルブロミド(0.32mL,2.7mmol)の脱水THF溶液(10mL)を加えた後、室温で19時間撹拌した。反応液を水(50mL)中にあけ、酢酸エチル(30ml×5)で抽出した。有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥後濃縮した。残留物をシリカゲルカラムクロマトグラフィー(溶出液 n−ヘキサン:酢酸エチル=1:1 v/v)で精製し、0.66g(86%)の表題化合物を無色油状物質として得た。
MS(FAB,(M+H)+) m/z 291 C12H16FO5P
【0076】
メチル 2−(ジメトキシホスホリル)−3−(3−フルオロフェニル)プロパノアートの合成
【化42】
〔化41〕の化合物と同様にして、表題化合物を得た。
MS(FAB,(M+H)+) m/z 291 C12H16FO5P
【0077】
メチル 2−(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)アクリラートの合成
【化43】
カリウムtert−ブトキシド(0.15g,1.34mmol)の脱水THF(20mL)溶液に0℃、アルゴン雰囲気下、化合物メチル 2−(ジメトキシホスホリル)−3−(2−フルオロフェニル)プロパノアート(〔化41〕の化合物)(0.30g,1.03mmol)の脱水THF(4mL)溶液を加え、30分撹拌した。次にアルデヒド誘導体(〔化40〕の化合物)(0.45g,1.03mmol)の脱水THF溶液(4mL)を加えた後、室温で16時間撹拌した。反応液を水(50mL)中にあけ、酢酸エチル(30ml×5)で抽出した。有機層を飽和食塩水で、洗浄後無水硫酸マグネシウムで乾燥後濃縮した。残留物をシリカゲルカラムクロマトグラフィー(溶出液 n−ヘキサン:酢酸エチル=4:1 v/v)で精製し、0.42g(69%)の表題化合物を無色油状物質として得た。
MS(FAB,(M+H)+) m/z 597 C38H44FNO4
【0078】
メチル (2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパノアートの合成
【化44】
メチル 2−(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)アクリラート(0.42g,0.70mmol) の酢酸エチル(30mL)溶液に、10%パラジウム担持活性炭(50mg)を加え、室温、200−300kPa水素加圧化2時間水素添加した。触媒をセライトで濾取し、酢酸エチルで洗浄した。濾液を濃縮し、残留物をシリカゲルカラムクロマトグラフィー(溶出液、n−ヘキサン:酢酸エチル=4:1 v/v)で精製し、0.37g(89%)の表題化合物を無色油状物質として得た。
1H NMR(300MHz,CDCl3)=δ7.70(d,J=8.7Hz,2H),7.40(d,J=8.7Hz,2H),7.24−7.10(m,3H),7.05−6.95(m,3H),4.61(d,J=5.7Hz,2H),3.96(t,J=6.3Hz,2H),3.50(s,3H),3.00−2.89(m,4H),2,74(dd,J=13.2,4.8Hz,1H),2.10(s,3H),1.91−1.70(m,14H),1.06(t,J=7.2Hz,3H)
MS (FAB,(M+H)+) m/z 598 C38H44FNO4
【0079】
(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸(〔化35〕の化合物)の最終合成過程
上記合成のメチル (2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパノアート(0.37g,0.62mmol) のエタノール溶液に、2mol/L水酸化ナトリウム水溶液(3.1mL,6.2mmol)を加え60℃で48時間撹拌した。反応液を濃縮し10%塩酸を溶液がpH1になるまで加え撹拌後析出した固体を吸引濾過した。得られた固体を酢酸エチルで溶かし無水硫酸マグネシウムで乾燥した後濃縮した。残留物をシリカゲルカラムクロマトグラフィー (溶出液 n−ヘキサン:酢酸エチル=2:1 v/v)で精製し、0.32g(89%) の表題化合物(〔実施例7〕の目的化合物)を白色固体として得た。
m.p.158−159℃
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.7Hz,2H),7.39(d,J=8.4Hz,2H),7.21−7.15(m,3H),7.06−6.95(m,3H),6.77−6.72(m,2H),4.57(d,J=5.4Hz,2H),3.94(t,J=6.5Hz,2H),2.98−2.80(m,4H),2.76(dd,J=13.8,4.8Hz,1H),2.10(s,3H),1.90−1.71(m,14H),1.06(t,J=7.4Hz,3H)
MS (FAB, M+H)+) m/z 584 C37H42NO4F
【0080】
〔実施例8〕(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸〔化45〕の合成例
【化45】
【0081】
メチル 2−(3−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)アクリラートの合成
【化46】
〔化43〕の合成操作と同様にして、表題化合物を得た。
MS(FAB,(M+H)+) m/z 597 C38H44FNO4
【0082】
メチル (3−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパノアートの合成
【化47】
〔化44〕の合成操作と同様にして、表題化合物を無色油状物質として得た。
1H NMR(300MHz,CDCl3)δ7.70(d,J=8.7Hz,2H),7.40(d,J=8.7Hz,2H),7.24−7.17(m,1H),7.10(s,1H),7.01(d,J=8.4Hz,1H),6.92−6.76(m,4H),6.66(t,J=5.7Hz,1H),4.61(d,J=5.7Hz,2H),3.96(t,J=6.5Hz,2H),3.51(s,3H),2.93−2.86(m,3H),2.80−2.69(m,2H),2.10(s,3H),1.90−1.72(m,14H),1.07(t,J=7.5Hz,3H).
MS (FAB,(M+H)+) m/z 598 C38H44FNO4
【0083】
次に、実施例7の化合物と同様にして、実施例8の化合物(〔化45〕)を得た。
m.p.201−202℃
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.4Hz,2H),7.39(d,J=8.1Hz,2H),7.23−7.15(m,2H),7.02(dd,J=2.1,8.1Hz,1H),6.94−6.85(m,3H),6.78−6.71(m,2H),4.58(d,J=5.4Hz,2H),3.95(t,J=6.6Hz,2H),2.99−2.87(m,3H),2.79−2.72(m,2H),2.09(s,3H),1.90−1.71(m,14H),1.06(t,J=7.35Hz,3H).
MS (FAB,(M+H)+) m/z 584 C37H42FNO4
【0084】
〔実施例9〕(4−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸(〔化48〕の化合物)の合成例
【化48】
【0085】
メチル−(4−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパノアートの合成
【化49】
〔化47〕の合成操作と同様にして、表題化合物を無色油状物質として得た。
1H NMR(300MHz,CDCl3)δ7.70(d,J=8.4Hz,2H),7.40(d,J=8.4Hz,2H),7.12−7.06(m,3H),7.00(dd,J=8.3,2.3Hz,1H),6.93(t,J=8.7Hz,2H),6.77(d,J=8.4Hz,1H),6.66(t,J=5.6Hz,1H),4.61(d,J=5.7Hz,2H),3.96(t,J=6.5Hz,2H),3.49(s,3H),2.93−2.84(m,3H),2.78−2.66(m,2H),2.10(s,3H),1.91−1.72(m,14H),1.06(t,J=7.4Hz,3H).
MS (FAB,(M+H)+) m/z 598 C38H44FNO4
【0086】
次に、実施例7の化合物と同様にして、実施例9の化合物(〔化48〕)を得た。
m.p.167−168℃
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.4Hz,2H),7.39(d,J=8.4Hz,2H),7.16−7.09(m,3H),7.02(dd,J=8.4,2.1Hz,1H),6.92(t,J=8.4Hz,2H),6.77−6.73(m,2H),4.58(d,J=6.0Hz,2H),3.95(t,J=6.5Hz 2H),2.96−2.85(m,3H),2.76−2.68(m,2H),2.10(s,3H),1.90−1.71(m,14H),1.06(t,J=7.4Hz,3H).
MS (FAB,(M+H)+) m/z 584 C37H42NO4F
【0087】
〔実施例10〕2−ベンジル−3−(2−フルオロ−5−((2−フルオロ−4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化50】
実施例7と同様の操作により、実施例10の化合物(〔化50〕)を得た。
1H−NMR(500MHz,CDCl3)δ8.00(t,J=8.5Hz,1H),7.35(m,1H),7.25−7.15(m,7H),7.05(dd,J=14.6,1.8Hz,1H),6.54(d,J=11.6Hz,1H),4.55(m,2H),3.91(t,J=6.4Hz,2H),3.02−2.96(m,2H),2.86−2.77(m,3H),2.10(s,3H),1.87−1.72(m,14H), 1.06(t,J=7.3Hz,3H);
MS (FAB) 602 (M+H)+
【0088】
〔実施例11〕2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸の合成例
【化51】
実施例7の同様の操作により、実施例11の化合物(〔化51〕)を得た。
1H−NMR(500MHz,CDCl3)δ8.02(t,J=8.5Hz,1H),7.34(m,1H),7.24−7.14(m,7H),7.04(dd,J=14.6,1.8Hz,1H),6.54(d,J=12.2Hz,1H),4.55(s,2H),3.91(t,J=6.7Hz,2H),2.86(m,2H),2.71(m,2H), 2.60(m,1H),2.09(s,3H),2.00−1.72(m,16H),1.06(t,J=7.3Hz,3H);
MS (FAB) 616 (M+H)+
【0089】
〔実施例12〕2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸の合成例
【化52】
実施例7と同様の操作により、実施例12の化合物(〔化52〕)を得た。
1H−NMR(500MHz,CDCl3)δ8.02(t,J=8.5Hz,1H),7.33(m,1H),7.28−7.12(m,7H),7.04(dd,J=14.6,1.8Hz,1H),6.54(d,J=11.6Hz,1H),4.55(d,J=5.5Hz,2H),3.91(t,J=6.7Hz,2H),2.85−2.77(m,2H),2.67−2.57(m,3H),2.10(s,3H),1.92−1.54(m,18H),1.07(t,J=7.3Hz,3H);
MS (FAB) 630 (M+H)+
【0090】
〔実施例13〕(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(ピリミジン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸の合成例
【化53】
実施例1と同様の操作により、実施例13の化合物〔化53〕を得た。
1H−NMR(400MHz,DMSO−d6)δ12.06(s,1H),8.94(d,J=4.8Hz,2H),8.91(t,J=5.8Hz,1H),8.47(d,J=8.8Hz,2H),8.04(d,J=8.4Hz,2H),7.49(t,J=4.8Hz,1H),7.19−7.16(m,2H),7.12−7.09(m,3H),7.02−7.01(m,2H),6.87(d,J=9.2Hz,1H),4.50−4.40(m,2H),3.93(t,J=6.4Hz,2H),2.82−2.71(m,3H),2.70−2.58(m,2H),1.77−1.69(m,2H),0.98(t,J=7.4Hz,3H)
【0091】
〔実施例14〕(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(チオフェン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸の合成例
【化54】
実施例1と同様の操作により、実施例14の化合物〔化54〕を得た。
1H−NMR(400MHz,DMSO−d6)δ12.05(s,1H),8.81(t,J=5.6Hz,1H),7.94(t,J=8.8Hz,2H),7.76(d,J=8.4Hz,2H),7.64(dd,J=3.6,1.2Hz,1H),7.62(dd,J=5.2,1.2Hz,1H),7.19−7.15(m,3H),7.12−7.09(m,3H),7.02−6.99(m,2H),6.86(d,J=8.4Hz,1H),4.49−4.38(m,2H),3.92(t,J=6.2Hz,2H),2.81−2.71(m,3H),2.68−2.56(m,2H),1.77−1.68(m,2H),0.98(t,J=7.4Hz,3H)
【0092】
〔実施例15〕(R)−2−ベンジル−3−(3−((4−(ピペリジン−1−イル)ベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化55】
実施例1と同様の操作により、実施例15の化合物〔化55〕を得た。
1H−NMR(400MHz,DMSO−d6)δ12.05(s,1H),8.46(t,J=5.8Hz,1H),7.76(d,J=8.8Hz,2H),7.18−7.08(m,5H),6.99(dd,J=8.4,2.0Hz,1H),6.95−6.92(m,3H),6.84(d,J=8.4Hz,1H),4.44−4.33(m,2H),3.91(t,J=6.4Hz,2H),3.27(t,J=5.2Hz,4H),2.77−2.69(m,3H),2.65−2.52(m,2H),1.77−1.68(m,2H),1.57(s,6H),0.98(t,J=7.4Hz,3H)
【0093】
〔実施例16〕(R)−2−ベンジル−3−(3−((2’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化56】
実施例1と同様の操作により、実施例16の化合物〔化56〕を得た。
1H−NMR(400MHz,CDCl3)δ7.80(d,J=8.4Hz,2H),7.59(dd,J=8.4,1.6Hz,2H),7.43(dt,J=7.6,1.8Hz,1H),7.37−7.32(m,1H),7.28−7.14(m,8H),6.80−6.76(m,2H),4.65−4.56(m,2H),3.95(t,J=6.6Hz,2H),3.01−2.87(m,3H),2.82−2.73(m,2H),1.88−1.79(m,2H),1.06(t,J=7.4Hz,3H)
【0094】
〔実施例17〕 (R)−2−ベンジル−3−(3−((3’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化57】
実施例1と同様の操作により、実施例17の化合物〔化57〕を得た。
1H−NMR(400MHz,DMSO−d6)δ8.87(t,J=5.6Hz,1H),8.00(d,J=8.4,2H),7.82(d,J=8.8Hz,2H),7.61−7.58(m,2H),7.55−7.49(m,1H),7.23(dt,J=8.8,1.6Hz,1H),7.19−7.15(m,2H),7.12−7.09(m,3H),7.03−7.00(m,2H),6.86(d,J=8.4Hz,1H),4.50−4.40(m,2H),3.92(t,J=6.4Hz,2H),2.79−2.71(m,3H),2.69−2.56(m,2H),1.77−1.69(m,2H),0.99(t,J=7.4Hz,3H)
【0095】
〔実施例18〕(R)−2−ベンジル−3−(3−((2’6’フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化58】
実施例1と同様の操作により、実施例18の化合物〔化58〕を得た。
1H−NMR(400MHz,DMSO−d6)δ12.06(s,1H),7.95(d,J=8.4Hz,2H),7.57(d,J=8.4,2H),7.53−7.46(m,1H),7.25(t,J=8.2Hz,2H),7.17−7.14(m,2H),7.11−7.09(m,3H),7.03−7.00(m,2H),6.87(d,J=8.4Hz,1H),4.51−4.40(m,2H),3.93(t,J=6.4Hz,2H),2.81−2.69(m,3H),2.68−2.57(m,2H),1.78−1.69
(m,2H),0.99(t,J=7.4Hz,3H)
【0096】
〔実施例19〕 (R)−2−ベンジル−3−(3−(2’−メチルビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化59】
実施例1と同様の操作により、実施例19の化合物〔化59〕を得た。
1H−NMR(400MHz,CDCl3)δ7.78(d,J=8.4Hz,2H),7.35(d,J=8.4Hz,2H),7.28−7.22(m,5H),7.20−7.17(m,5H),7.04(dd,J=8.4,2.2Hz,1H),6.81−6.76(m,2H),4.65−4.56(m,2H),3.96(t,J=6.4Hz,2H),3.02−2.87(m,3H),2.82−2.73(m,2H),2.24(s,3H),1.88−1.80(m,2H),1.06(t,J=7.4Hz,3H)
【0097】
〔実施例20〕 (R)−2−ベンジル−3−(3−(ビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化60】
実施例1と同様の操作により、実施例20の化合物〔化60〕を得た。
1H−NMR(400MHz,CDCl3)δ7.80(d,J=8.4Hz,2H),7.63−7.57(m,4H),7.45(t,J=7.4Hz,2H),7.37(t,J =7.4Hz,1H),7.27−7.23(m,2H),7.04(dd,J=8.4,2.4Hz,1H),6.81(t,J=5.8Hz,1H),6.77(d,J=8.0Hz,1H),4.65−4.55(m,2H),3.95(t,J=6.4Hz,2H),3.02−2.86(m,3H),2.80−2.73(m,2H),1.88−1.79(m,2H),1.05(t,J=7.4Hz,3H)
【0098】
〔実験例〕
1.本発明の化合物のPPARγアゴニスト活性の測定
10%脱脂牛血清を含むダルベッコ変法イ−グル培地(FCS/DMEM)にて培養したヒト胎児腎細胞(HEK293)に、酵母の転写因子(GAL4)のDNA結合領域とヒト型PPARの各サブタイプ(α、δ、γ)のリガンド結合領域との融合蛋白質を発現する受容体プラスミド及びそのレポータープラスミド、さらに内部標準用のβガラクトシダーゼプラスミドをリン酸カルシウム法にて無血清状態にてコトランスフェクションした。その後、被検化合物を添加して16時間後にルシフェラ−ゼ活性ならびにβガラクトシダーゼ活性を測定し、内部標準により補正した。
上記方法により測定したPPAR(実施例1〜12)に対するEC50の値を表1に示す。
【表1】
【0099】
また、一般式(I)のR1をアダマンチル基以外の置換基にして、水溶性の向上を試みた(実施例13〜20)。この結果を表2に示す。
【表2】
【0100】
表1に示すように、本発明の化合物は、PPARγに選択的に作用するアゴニスト活性を有する。これらのEC50は、同様の方法で測定した既知のPPARγ選択的なアゴニストであるロシグリタゾン(EC50=43nM)やPPARのパンアゴニストであるTIPP703((S)−2−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシベンジル)ブタン酸、EC50=37nM;Bioorg Med Chem Lett. 2008, 18(3), 1110-1115に記載の化合物番号12)と比較しても、同等以上の値を示しており、良好なPPARγ選択的アゴニストであることが明らかとなった。
また、一般式(I)のR1をアダマンチル基以外の置換基にすると、EC50は、若干悪くなるものもあるが、水溶性は、R1がアダマンチル基である化合物(例えば、実施例1の化合物)に比較して、改善されている。特に、実施例13、15及び18の化合物は、実施例1の化合物よりも水溶性が向上し、かつ、EC50の値についても、既知のPPARγ選択的なアゴニストであるロシグリタゾンと同等以上の値を示している。
【0101】
2.PPARγアゴニストの癌細胞への影響
次に、本発明の化合物が癌細胞に及ぼす影響について検討を行った。本実施例では、典型的なスキルス胃癌の臨床経過を呈するスキルス胃癌細胞株OCUM−2MD3(大阪市立大学医学部の八代正和先生から供与を受けた)を使用して実験を行った。また、正常細胞に対する化合物の影響をみるために、正常細胞株であるOUMS−24(Bai L, et al., Int J Cancer. 1993 ; 53: 451-456)を使用して実験を行った。
OCUM−2MD3又はOUMS−24を6ウェルプレートに1.5×104細胞/ウェル(培地:DMEM+10% FBS)となるように播き、翌日(Day1)、本発明の化合物(実施例1の化合物、実施例13の化合物及び実施例15の化合物)、トログリタゾン及びDMSOのみで処理を行い、4日目(Day4)に化合物を含む培地で培地交換を行い、5日目(Day5)に細胞数の計測を行った(5視野にて計測を行った)。また、アポトーシス細胞の割合は、(トリパンブルー染色細胞数)/(総細胞数)により算出した。
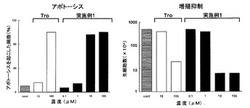
図1に実施例1の化合物によるアポトーシスの誘導効果及び細胞増殖抑制効果に関する計測結果を示す。実施例1の化合物を、0.1μM、1μM、10μM及び100μMとなるように添加し、コントロールとしてトログリタゾンを10μM及び100μMとなるように添加した。トログリタゾンが100μMで活性(アポトーシス誘導活性及び細胞増殖抑制活性)を示すのに対し、実施例1の化合物はその1/10である10μMでアポトーシス誘導効果及び細胞増殖抑制効果を示した。
【0102】
次に、癌細胞ではない正常な細胞に対し、本発明の化合物が与える影響について検討を行った。図2は、本発明の化合物(実施例1の化合物、実施例13の化合物及び実施例15の化合物)のスキルス胃癌細胞(OCUM−2MD)及び正常細胞(OUMS−24)に対するアポトーシス誘導効果について、検討を行った結果である。実施例1の化合物については、その濃度依存的にOCUM−2MDのアポトーシスを誘導したのに対し、OUMS−24については、少なくとも10μMまでは、アポトーシスを誘導しなかった。一方、実施例13及び実施例15の化合物については、100μMにおいても、OCUM−2MDに選択的にアポトーシスを誘導した。
以上の結果から、本発明の化合物は、トログリタゾンよりも低用量で癌細胞に対する増殖抑制効果を示し(図1)、かつ、正常細胞の増殖にはほとんど影響を与えない(図2)ことが分かる。
【産業上の利用可能性】
【0103】
本発明の化合物は、PPARγのアゴニストの活性を有する。従って、これらの化合物を有効成分として含有する医薬は、PPARγの活性異常に起因する疾患の治療等に効果を発揮することが期待される。
【技術分野】
【0001】
本発明は、新規α−置換プロピオン酸誘導体、及び、該α−置換プロピオン酸誘導体であって、ペルオキシゾーム増殖薬活性化受容体(PPAR)γに選択的なアゴニストに関し、さらに、PPARγ活性の異常に起因する疾患等の治療及び/又は予防のための医薬又は医薬組成物、並びに、PPARγ活性の異常に起因する疾患等の治療及び/又は予防方法に関する。
【背景技術】
【0002】
ペルオキシソーム増殖剤応答性受容体(Peroxisome proliferator−activated receptor:PPAR、以下PPARとする)は、核内受容体スーパーファミリーに属するリガンド依存性の転写因子であり、標的遺伝子の転写をリガンド依存的に誘導する。すなわち、リガンドがPPARに結合すると、PPARは標的遺伝子のプロモーター領域に存在するPPAR応答配列(PPAR responsive element:PPRE)に結合し、標的遺伝子の転写が誘導される。
細胞内において、PPARはレチノイドXレセプター(RXR)とヘテロ二量体を形成する。このヘテロ二量体がPPAR応答配列(PPRE)として知られるDNA配列に結合して、各種遺伝子の転写を活性化する。また、PPAR/RXRヘテロ二量体は、DRIP−205やSRC−1などの活性化補助因子を取り込んで、標的遺伝子にコードされるmRNAの発現レベルを調節する。
【0003】
これまでに組織分布を異にする3種類のサブタイプ(α型、β/δ型、γ型)がヒトをはじめとする様々な動物種で同定されている。これらのうち、PPARαは、脂肪酸の異化能の高い肝臓、腎臓、心臓及び筋肉等に分布しており、特に肝臓において高発現が認められ、PPARαによって標的遺伝子の転写が誘導されると、血中中性脂肪の低下、HDLコレステロールの増加、体重の減少、血管新生の促進等が誘起される。PPARβ/δは、骨格筋を中心に生体内各組織に普遍的に発現しており、PPARβ/δの活性化により、骨格筋における脂肪酸の異化、HDLコレステロールの増加、インスリン抵抗性の改善、肥満の抑制などが誘導されることが明らかとなってきている(非特許文献1、非特許文献2)。
【0004】
PPARγは、脂肪細胞やマクロファージに高発現しており、脂肪細胞の分化、インスリン感受性の獲得などに関与するタンパク質を誘導する。アイソフォームとして、PPARγ1、γ2及びγ3の少なくとも3種類の存在が確認されており、これらは、選択的スプライシングの結果、発現すると考えられている。また、PPARγは、大腸癌、乳癌、前立腺癌、膵臓癌、肺癌、骨髄性白血病細胞及びリンパ性白血病細胞などのおおくの癌細胞においても、その発現が確認されている(非特許文献3〜5)。さらに、PPARγのリガンドであるチアゾリジンジオン(TZDs)が種々の異なるタイプの腫瘍細胞(胃腸、胆管及び膵臓の腺癌)の増殖を阻害することが報告されており(非特許文献4)、PPARγが抗癌剤のターゲットとして、にわかに注目をあびつつある。PPARγによる抗腫瘍機構については、種々の組織由来の腫瘍毎に異なることも予想され、その全容は未だ明らかにされていない。ただ、PPARγが、サイクリンD1の分解を促進しG1期で細胞増殖を停止させること(非特許文献6)、E2F−DPのDNA結合を阻害してその転写活性を抑制する(非特許文献7)などの報告から、PPARγが細胞増殖の抑制に直接関与していることが示唆されている。さらに、PPARγの活性が上昇すると血管新生因子であるVEGFの産生が減少することなども報告されており、PPARγの血管新生阻害作用も考慮されている(非特許文献8)。
【0005】
PPARの各サブタイプは、各々のターゲット遺伝子の転写を誘導することにより、脂肪代謝、インスリン抵抗の改善など、いわゆる、メタボリック症候群として知られる諸症状の緩和、あるいは、癌細胞の増殖抑制などに寄与していることが予想されている。すでに、PPARαに対する外因性リガンドとして、フェノフィブラート、ベザフィブラート、クロフィブラートなどのいわゆるフィブラート系の薬剤が、また、PPARγに対する外因性リガンドとして、トログリタゾン、ピオグリタゾン、ロシグリタゾンなどのいわゆるチアゾリジン系の薬剤が知られている。また、フィブラート系、チアゾリジン系以外で、PPARの各アイソフォームをターゲットとする化合物もいくつか報告されている(特許文献1〜6を参照のこと)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】WO01/092201号
【特許文献2】WO00/75103号
【特許文献3】WO2004/056748号
【特許文献4】WO2004/046091号
【特許文献5】WO03/051821号
【特許文献6】WO02/098840号
【非特許文献】
【0007】
【非特許文献1】Oliverら、Proc.Natl.Acad.Sci.USA. 98:5306−5311 2001
【非特許文献2】Tanakaら、Proc.Natl.Acad.Sci.USA. 100:15924−15929 2003
【非特許文献3】Smithら、Invest New Drugs.20:195−200 2002
【非特許文献4】Tsujieら、Exp.Cell Res.289:143−151 2003
【非特許文献5】Campbellら、PPAR Research Volume 2008,Article ID 314974
【非特許文献6】Qinら、Cancer Res.63 958−964 2003
【非特許文献7】Altiokら、Genes Dev.11 1987−1988 1997
【非特許文献8】Panigraphyら、J.Clin.Invest.110 923−932 2002
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は、新規α−置換プロピオン酸誘導体、及び該α−置換プロピオン酸誘導体であってPPARγに選択的なアゴニストの提供を目的とする。
また、本発明は、該α−置換プロピオン酸誘導体を有効成分として含むPPARγ転写活性化剤の提供を目的とする。
さらに、本発明は、該α−置換プロピオン酸誘導体を有効成分として含む、PPARγ転写活性の異常に起因する疾患(例えば、癌など)の治療剤及び/又は予防剤の提供を目的とする。
【課題を解決するための手段】
【0009】
上述の通り、脂肪代謝異常などのメタボリック症候群や癌の治療薬として注目を浴びているPPARアゴニストであるが、解決すべき問題も存在している。既存のPPARアゴニスト(例えば、PPARγアゴニスト)の中には、その強力なアゴニスト活性に基づく副作用(例えば、肝機能障害、浮腫、体重増加など)を誘発する化合物が報告されている(特開2007−314464明細書などを参照)。そのため、現段階においても、より薬理効果が高く、副作用(絶対的又は相対的に)等の少ないPPARアゴニスト及びこれを含む治療薬の開発が望まれている。
本発明者らは、上記事情に鑑み、鋭意研究を行ったところ、新規α−置換プロピオン酸誘導体を合成し、該α−置換プロピオン酸誘導体であってPPARγに選択的な高いアゴニスト活性を示す化合物を見出し、本発明を完成させた。
【0010】
すなわち本発明は、 一般式(I):
【化1】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、nは0から10の整数を表す]で表されるα−置換フェニルプロピオン酸誘導体若しくはその薬学上許容される塩又は水和物である。
さらに、本発明は上記一般式(I)で表されるα−置換フェニルプロピオン酸誘導体であるPPARγに選択的なアゴニストである。
【0011】
また、本発明は、上記一般式(I)で表されるα−置換フェニルプロピオン酸誘導体若しくはその薬学上許容される塩又は水和物を含有する、PPARγ転写活性化剤である。
【0012】
さらに、本発明は、上記一般式(I)で表されるα−置換フェニルプロピオン酸誘導体若しくはその薬学上許容される塩又は水和物を含有する医薬である。
【0013】
また、本発明は、上記一般式(I)で表される化合物の製造に適する、以下の一般式(II)及び一般式(III)の化合物である。
【化2】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基を表す]
【化3】
[式中、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R4は水素原子又はハロゲン原子、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、Pは、不斉補助基又は炭素数1から10の直鎖状又は分岐状アルキル基、nは0から10の整数を表す]
【発明の効果】
【0014】
本発明により、新規α−置換フェニルプロピオン酸誘導体及び該α−置換フェニルプロピオン酸誘導体であってPPARγに選択的なアゴニストが提供される。また、本発明により、PPARγ転写活性化剤が提供される。
【0015】
さらに、本発明により、PPARγ転写活性の異常によって惹起される疾患、例えば、癌、メタボリック症候群(例えば、糖尿病、高脂血症、高コレステロール症、高血圧症、肥満症、動脈硬化症など)などの予防又は治療剤が提供される。
【0016】
また、本発明の一般式(I)で示される化合物は、一般式(II)又は一般式(III)の中間体化合物を使用することで、迅速かつ効率的な製造が可能となる。
【図面の簡単な説明】
【0017】
【図1】本発明の化合物の癌細胞の増殖への効果。 本発明の化合物(実施例1の化合物)の癌細胞(OCUM−2MD3)に対するアポトーシス誘導効果(アポトーシス)及び増殖抑制効果(増殖抑制)を示す。実施例1の化合物は、0.1、1、10、100μM使用した。対照として、DMSOのみ(cont)、あるいは、トログリタゾン(Tro;10μM、100μM)を添加して実験を行った。
【図2】本発明の化合物の正常細胞への影響。 本発明の化合物(実施例1、実施例13及び実施例15の化合物)の癌細胞(OCUM−2MD3)及び正常細胞(OUMS−24)に対するアポトーシス誘導率を示す。
【発明を実施するための形態】
【0018】
本発明の実施形態の1つは、一般式(I)で表されるα−置換プロピオン酸誘導体若しくはその薬学上許容される塩又は水和物、あるいは、該α−置換プロピオン酸誘導体であってPPARγに選択的なアゴニストである。ここで、「アゴニスト」とは、PPARγに結合して、PPARγによる標的遺伝子の転写活性化を誘導する化合物のことである。
一般式(I)において、R1は、アダマンチル基、無置換又は置換基を有していても良いフェニル基、無置換又は置換基を有していても良いピリジル基、無置換又は置換基を有していても良いピリミジニル基、無置換又は置換基を有していても良いピラジニル基、無置換又は置換基を有していても良いピペリジン基、無置換又は置換基を有していても良いアゼピル基、無置換又は置換基を有していても良いピペラジニル基であり、あるいは、2−チエニル基、モルホリノ基である。ここで、置換基を有するフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基、ピペラジニル基の置換基としては、例えば、ハロゲン原子が好ましく、特に、フッ素原子が好ましい。
R2及びR4は、同一又は相異なって、水素原子又はハロゲン原子であり、好ましくは、水素原子又はフッ素原子である。
R3は炭素数1から10の直鎖状又は分岐状アルキル基、好ましくは、炭素数1〜5の直鎖状アルキル基、より好ましくは、プロピル基である。
R5は置換基を有していても良いフェニル基又はシクロヘキシル基であり、置換基を有するフェニル基又はシクロヘキシル基の置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基であり、好ましくは、フェニル基、フルオロフェニル基、シクロヘキシル基であり、より好ましくは、フェニル基又はフルオロフェニル基である。R5の置換基の位置は任意であるが、例えば、2位、4位などが好ましい。nは0〜10の整数を表し、好ましくは、1〜3の整数である。
【0019】
一般式(I)で表される本発明の化合物は、一般式(I)で表されるα−置換プロピオン酸誘導体のみならず、その塩又はそれらの溶媒和物若しくは水和物であっても良い。α−置換プロピオン酸誘導体の塩は特に限定されるものではなく、慣用の塩、例えば、ナトリウム塩、カリウム塩、リチウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アルミニウム塩等の金属塩が挙げられ、好ましくは薬学上許容されるものである。
また、一般式(I)で表されるα−置換プロピオン酸誘導体には、特に断らない限り、その互変異性体、幾何異性体(例えば、E体、Z体など)、鏡像異性体等の立体異性体も含まれる。すなわち、一般式(I)で表されるα−置換プロピオン酸誘導体中に、1個又は2個以上の不斉炭素が含まれる場合、不斉炭素の立体化学については、それぞれ独立して(R)体又は(S)体のいずれかをとることができ、該誘導体の鏡像異性体又はジアステレオ異性体などの立体異性体として存在することがある。
本発明の医薬の有効成分としては、純粋な形態の任意の立体異性体、立体異性体の任意の混合物、ラセミ体などを用いることが可能である。
【0020】
また、本発明の他の実施形態は、一般式(II)又は一般式(III)で示される化合物である。一般式(II)又は一般式(III)で示される化合物は、一般式(I)のα−置換プロピオン酸誘導体を製造するのに適した化合物である。
【0021】
一般式(II)において、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、無置換又は置換基を有していても良いピリジル基、無置換又は置換基を有していても良いピリミジニル基、無置換又は置換基を有していても良いピラジニル基、無置換又は置換基を有していても良いピペリジン基、無置換又は置換基を有していても良いアゼピル基、無置換又は置換基を有していても良いピペラジニル基であり、あるいは、2−チエニル基、モルホリノ基であり、置換基を有するフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基、ピペラジニル基の置換基としては、例えば、ハロゲン原子が好ましく、特に、フッ素原子が好ましい。好ましいR1として、アダマンチル基、ピリミジン−2−イル基、ピペリジニル基を挙げることができる。
R2は水素原子又はハロゲン原子であり、好ましくはハロゲン原子、特に好ましくはフッ素原子である。R3は炭素数1から10の直鎖状又は分岐状アルキル基、好ましくは、炭素数1〜5の直鎖状アルキル基、より好ましくは、プロピル基である。R4は水素原子又はハロゲン原子を表し、好ましくは水素原子又はフッ素原子である。
【0022】
一般式(III)において、R3は炭素数1から10の直鎖状又は分岐状アルキル基、好ましくは、炭素数1〜5の直鎖状アルキル基、より好ましくは、プロピル基である。R4は水素原子又はハロゲン原子を表し、好ましくは、水素原子又はフッ素原子である。R5は置換基を有していても良いフェニル基又はシクロヘキシル基であり、置換基を有するフェニル基又はシクロヘキシル基の置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基であり、好ましくは、フェニル基、フルオロフェニル基、シクロヘキシル基であり、より好ましくは、フェニル基又はフルオロフェニル基である。R5の置換基の位置は任意であるが、例えば、2位、4位などが好ましい。nは0〜10の整数を表し、好ましくは、1〜3の整数である。Pは、不斉補助基である。不斉補助基としては、反応を妨げない限り既知のものを適宜使用することができる。例えば、4−ベンジルオキサゾリジノン、4−イソプロピルオキサゾリジノン等を挙げることができる。
【0023】
一般式(I)で示されるPPARγに選択的なアゴニスト及びその塩としては、限定はしないが、例えば、次のものが挙げられる。
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸、
(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸、
(S)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸、
(R)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸、
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸、
(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸、
(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
(3−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
(4−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
2−ベンジル−3−(2−フルオロ−5−((2−フルオロ−4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸、
2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸。
(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(ピリミジン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸
(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(チオフェン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸
(R)−2−ベンジル−3−(3−((4−(ピペリジン−1−イル)ベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−((2’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−((3’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−((2’6’ジフルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−(2’−メチルビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−(ビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
【0024】
本発明の一般式(I)で表される化合物のうち、R4が水素原子、R5が無置換フェニル基である光学活性な化合物(Ia)は、以下の方法により製造することができる(スキーム1)。
【化4】
【0025】
すなわち、一般式(Ia):
【化5】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、nは0から10の整数を表す]
で表される化合物は、以下の:
【化6】
[式中、R3は前記と同義である]で表される化合物(A)と、以下の:
【化7】
[式中、nは前記と同義である]で表される化合物(G)とを反応させ(第一工程)、得られた以下の:
【化8】
[式中、R3及びnは前記と同義である]で表される化合物(B)を還元する(第二工程)ことにより製造することができる以下の:
【化9】
[式中、R3及びnは前記と同義である]で表される化合物(C)を還元する(第三工程)ことにより製造することができる以下の:
【化10】
[式中、R3及びnは前記と同義である]で表される化合物(D)を酸化する(第四工程)ことにより製造することができる以下の:
【化11】
[式中、R3及びnは前記と同義である]で表される化合物(E)と以下の:
【化12】
[式中、R1及びR2は前記と同義である]で表わされる化合物(H)を還元的アミドアルキル化(第五工程)ことにより製造することができる以下の:、
【化13】
[式中、R1、R2、R3およびnは前記と同義である]で表わされる化合物(F)の不斉補助基を脱保護する(第六工程)ことにより合成することができる。
【0026】
第一工程の反応は、テトラヒドロフランやジエチルエーテル、ヘキサン等の溶媒中塩基として例えば水素化ナトリウムのようなアルカリ金属水素化物、ブチルリチウムのような有機金属化合物、リチウムジイソプロピルアミド、ナトリウムビス(トリメチルシリル)アミドのような金属アミドを用いる事ができる。反応温度としては−100℃から室温にて、好適には−80℃から0℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0027】
第二工程の反応はパラジウム担持活性炭、白金担持活性炭、酸化白金、ロジウム担持アルミナ等の金属触媒存在下、エタノール、メタノール、テトラヒドロフラン、酢酸エチル、N,N−ジメチルホルムアミド等の溶媒中水素圧98.1kPaから491kPaで実施する事ができる。反応温度としては0℃から150℃にて、好適には室温から100℃にて実施する事ができる。反応時間は通常0.5〜24時間、好ましくは1〜5時間である。
【0028】
第三工程の反応は、テトラヒドロフランやジエチルエーテル、ヘキサン等の溶媒中塩基として例えばボランTHFコンプレックス、ジイソブチルアルミニウムヒドリド等の塩基を用いて実施する事ができる。反応温度としては−50℃から100℃にて、好適には−20℃から50℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0029】
第四工程の反応は、塩化メチレンやクロロホルム等の溶媒中酸化剤として例えば活性化二酸化マンガン、ピリジニウムクロロクロメート、ピリジニウムジクロメート等の塩基酸化剤を用いて実施する事ができる。反応温度としては−50℃から100℃にて、好適には−0℃から溶媒の還流温度にて実施する事ができる。反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0030】
第五工程の反応は、トルエン、ベンゼン、アセトニトリル、ジオキサン、N,N−ジメチルホルムアミド等の溶媒中、酸(例えば、トリフルオロ酢酸、トリフルオロメタンスルホン酸)の存在下、還元剤としてトリエチルシランの存在下で実施することができる。この際の反応温度は通常−20〜150℃、好ましくは0〜100℃であり、反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0031】
第六工程の反応は、アルカリ性条件下で実施する事ができる。アルカリ性条件としては水酸化リチウムと過酸化水素の混合物が用いられる。反応温度としては−20℃から100℃にて、好適には0℃から50℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0032】
また、本発明の一般式(I)で表される化合物のうち、R1がアダマンチル基、R2及びR4がフッ素原子、R3がプロピル基、R5が水素原子、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基を有するフェニル基又はシクロヘキシル基、nが1である化合物(Ib)は、以下の方法によって製造することができる(スキーム2)。
【化14】
【0033】
すなわち、一般式(Ib):
【化15】
[式中、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)を表す]で表される化合物は、以下の:
【化16】
で表される化合物(I)をホルミル化(第七工程)して得られる以下の:
【化17】
化合物(J)と以下の:
【化18】
化合物(O)とを還元的アミドアルキル化(第八工程)して得られる以下の:
【化19】
化合物(K)をホルミル化する(第九工程)ことにより製造することができる以下の:
【化20】
化合物(L)と以下の:
【化21】
[式中、R5は前記と同義である]で表される化合物(P)とを反応させる(第十工程)ことにより製造することができる以下の:
【化22】
[式中、R5は前記と同義である]で表される化合物(M)を還元する(第十一工程)ことにより製造することができる以下の:
【化23】
[式中、R5は前記と同義である]で表わされる化合物(N)を加水分解(第十二工程)することにより製造することができる。
【0034】
第七工程の反応は、ジクロロメタンやクロロホルム、四塩化炭素等の溶媒中ホルミル化剤としてジクロロメチルメチルエーテルを用い、さらにルイス酸として四塩化チタンやチタニウムテトライソプロポキシド、塩化アルミニウムを用いて製造する事ができる。反応温度としては−100℃から室温にて、好適には−80℃から0℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0035】
第八工程の反応は、トルエン、ベンゼン、アセトニトリル、ジオキサン、N,N−ジメチルホルムアミド等の溶媒中、酸(例えば、トリフルオロ酢酸、トリフルオロメタンスルホン酸)の存在下、還元剤としてトリエチルシランの存在下で実施することができる。この際の反応温度は通常−20〜150℃、好ましくは0〜100℃であり、反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0036】
第九工程の反応は、ジクロロメタンやクロロホルム、四塩化炭素等の溶媒中ホルミル化剤としてジクロロメチルメチルエーテルを用い、さらにルイス酸として四塩化チタンやチタニウムテトライソプロポキシド、塩化アルミニウムを用いて製造する事ができる。反応温度としては−100℃から室温にて、好適には−80℃から0℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0037】
第十工程の反応はテトラヒドロフラン、トルエン、ジオキサン、N,N−ジメチルホルムアミド等の溶媒中、塩基としては例えば水素化ナトリウムのようなアルカリ金属水素化物、ブチルリチウムのような有機金属化合物、リチウムジイソプロピルアミドのような金属アミド、ナトリウムメトキシドやカリウム t−ブトキシドのような金属アルコキシドを用いる事ができる。反応温度としては−20℃から150℃にて、好適には0℃から50℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0038】
第十一工程の反応は、パラジウム担持活性炭、白金担持活性炭、酸化白金、ロジウム担持アルミナ等の金属触媒存在下、エタノール、メタノール、テトラヒドロフラン、酢酸エチル、N,N−ジメチルホルムアミド等の溶媒中水素圧1kgf/cm2から5kgf/cm2で実施する事ができる。反応温度としては0℃から100℃にて、好適には室温から80℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは6〜24時間である。
【0039】
第十二工程の反応は、アルカリ性条件下で行う事ができる。アルカリ性条件としては水酸化リチウム、水酸化ナトリウム、水酸化カリウム等が用いられる。反応温度としては0℃から80℃にて、好適には室温から60℃にて実施する事ができる。反応時間は通常1〜48時間、好ましくは2〜24時間である。
【0040】
本発明には、一般式(I)で示されるα−置換プロピオン酸誘導体を含むPPARγ転写活性化剤(PPARγ標的遺伝子の転写を活性化(又は誘導)する薬剤)が含まれる。
【0041】
さらに、本発明のα−置換プロピオン酸誘導体であってPPARγに選択的なアゴニストを含む医薬も本発明の範囲に含まれる。
本発明の医薬は、PPARγの異常調節、特に、その活性が抑制されることによって発症する疾患等の治療剤又は予防剤として使用することができる。PPARγの既存のアゴニストであるチアゾリジン誘導体がインスリン抵抗性を改善し(例えば、Yamauchiら, J. Biol. Chem., 276:41245-41254, 2001など)、血糖値の調製に有効であることの他、血中遊離脂肪酸、TGの低下作用、HDL−C増加作用などが確認されており(例えば、Leeら, Endocrinology, 144:2201-2207, 2003など)、PPARγのアゴニストは糖尿病、高脂血症など、メタボリックシンドロームに関連する疾患の治療等に利用できることが知られている。さらに、PPARγは、大腸癌、乳癌、前立腺癌、膵臓癌、肺癌、骨髄性白血病細胞及びリンパ性白血病細胞などの多くの癌細胞においても、その発現が確認されている(非特許文献3〜5)。また、PPARγのアゴニストであるチアゾリジンジオン(TZDs)が種々の異なるタイプの腫瘍細胞(胃腸、胆管及び膵臓の腺癌)の増殖を阻害することなどから(非特許文献4)、PPARγのアゴニストは、抗癌剤としても使用することができる。
【0042】
一般式(I)で示されるPPARγに選択的なアゴニストを含む医薬は、脂質代謝異常、高脂血症、糖尿病、高血圧、高コレステロール症、肥満症、などのメタボリックシンドローム、並びに、メタボリックシンドロームを基盤として発症する、例えば、動脈硬化性疾患などの治療剤又は予防剤として使用することができる。さらに、一般式(I)で示されるPPARγに選択的なアゴニストを含む医薬は、癌の治療剤又は予防剤、例えば、大腸癌、乳癌、前立腺癌、膵臓癌、肺癌、骨髄性白血病細胞及びリンパ性白血病細胞の他、特に、胃腸、胆管及び膵臓などに発症する腺癌の治療剤又は予防剤として使用することができる。また、一般式(I)で示される化合物を含む本発明の医薬は、胃腺癌のうち印環細胞癌(進行型の低分化腺癌、非充実型(スキルス;Scirrhous)などを含む)などの治療にも使用することができる。
【0043】
本発明の医薬の有効成分としては、上記一般式(I)で表される化合物のほか、生理学的に許容されるその塩を用いても良い。塩としては、例えば、酸性基が存在する場合には、リチウム、ナトリウム、カリウム、マグネシウム、カルシウム等のアルカリ金属及びアルカリ土類金属塩;アンモニア、メチルアミン、ジメチルアミン、トリメチルアミン、ジシクロヘキシルアミン、トリス(ヒドロキシメチル)アミノメタン、N,N−ビス(ヒドロキシエチル)ピペラジン、2−アミノ−2−メチル−1−プロパノール、エタノールアミン、N−メチルグルカミン、L−グルカミン等のアミンの塩;又はリジン、δ−ヒドロキシリジン、アルギニンなどの塩基性アミノ酸との塩を形成することができる。塩基性基が存在する場合には、塩酸、臭化水素酸、硫酸、硝酸、リン酸等の鉱酸の塩;メタンスルホン酸、ベンゼンスルホン酸、パラトルエンスルホン酸、酢酸、プロピオン酸塩、酒石酸、フマル酸、マレイン酸、リンゴ酸、シュウ酸、コハク酸、クエン酸、安息香酸、マンデル酸、ケイ皮酸、乳酸、グリコール酸、グルクロン酸、アスコルビン酸、ニコチン酸、サリチル酸等の有機酸との塩;又はアスパラギン酸、グルタミン酸などの酸性アミノ酸との塩などを挙げることができる。
さらに、本発明の医薬の有効成分として、一般式(I)で表される化合物又はその塩の溶媒和物若しくは水和物を用いることもできる。
【0044】
本発明の医薬は、有効成分である一般式(I)で表される化合物及び薬理学的に許容されるその塩、又はそれらの溶媒和物若しくはそれらの水和物自体を投与しても良いが、一般的には、有効成分である一般式(I)の化合物と1又は2以上の製剤用添加物とを含む医薬組成物の形態で投与することが望ましい。本発明の医薬の有効成分としては、一般式(I)の化合物の2種以上を組み合わせて用いることができ、上記医薬組成物には、癌、糖尿病、高脂血症、高コレステロール症、高血圧症、肥満症、動脈硬化症などの諸症状の予防又は治療のための他の既知の有効成分を配合することも可能である。
【0045】
医薬組成物の種類は特に限定されず、剤型としては、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤、懸濁剤、座剤、軟膏、クリーム剤、ゲル剤、貼付剤、吸入剤、注射剤等が挙げられる。これらの製剤は常法に従って調製される。なお、液体製剤にあっては、用時、水又は他の適当な溶媒に溶解又は懸濁する形であっても良い。また錠剤、顆粒剤は周知の方法でコーティングしても良い。注射剤の場合には、本発明の化合物を水に溶解させて調製されるが、必要に応じて生理食塩水あるいはブドウ糖溶液に溶解させてもよく、また緩衝剤や保存剤を添加しても良い。経口投与用又は非経口投与用の任意の製剤形態で提供される。例えば、顆粒剤、細粒剤、散剤、硬カプセル剤、軟カプセル剤、シロップ剤、乳剤、懸濁剤又は液剤等の形態の経口投与用医薬組成物、静脈内投与用、筋肉内投与用、若しくは皮下投与用などの注射剤、点滴剤、経皮吸収剤、経粘膜吸収剤、点鼻剤、吸入剤、坐剤などの形態の非経口投与用医薬組成物として調製することができる。注射剤や点滴剤などは、凍結乾燥形態などの粉末状の剤形として調製し、用時に生理食塩水などの適宜の水性媒体に溶解して用いることもできる。また、高分子などで被覆した徐放製剤を脳内に直接投与することも可能である。
【0046】
医薬組成物の製造に用いられる製剤用添加物の種類、有効成分に対する製剤用添加物の割合、又は医薬組成物の製造方法は、組成物の形態に応じて当業者が適宜選択することが可能である。製剤用添加物としては無機又は有機物質あるいは固体又は液体の物質を用いることができ、一般的には、有効成分重量に対して1重量%から90重量%の間で配合することができる。具体的には、その様な物質の例として乳糖、ブドウ糖、マンニット、デキストリン、シクロデキストリン、デンプン、蔗糖、メタケイ酸アルミン酸マグネシウム、合成ケイ酸アルミニウム、カルボキシメチルセルロースナトリウム、ヒドロキシプロピルデンプン、カルボキシメチルセルロースカルシウム、イオン交換樹脂、メチルセルロース、ゼラチン、アラビアゴム、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、ポリビニルアルコール、軽質無水ケイ酸、ステアリン酸マグネシウム、タルク、トラガント、ベントナイト、ビーガム、酸化チタン、ソルビタン脂肪酸エステル、ラウリル硫酸ナトリウム、グリセリン、脂肪酸グリセリンエステル、精製ラノリン、グリセロゼラチン、ポリソルベート、マクロゴール、植物油、ロウ、流動パラフィン、白色ワセリン、フルオロカーボン、非イオン性界面活性剤、プロピレングルコール、水等が挙げられる。
【0047】
経口投与用の固形製剤を製造するには、有効成分と賦形剤成分例えば乳糖、澱粉、結晶セルロース、乳酸カルシウム、無水ケイ酸などと混合して散剤とするか、さらに必要に応じて白糖、ヒドロキシプロピルセルロース、ポリビニルピロリドンなどの結合剤、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウムなどの崩壊剤などを加えて湿式又は乾式造粒して顆粒剤とする。錠剤を製造するには、これらの散剤及び顆粒剤をそのまま或いはステアリン酸マグネシウム、タルクなどの滑沢剤を加えて打錠すれば良い。これらの顆粒又は錠剤はヒドロキシプロピルメチルセルロースフタレート、メタクリル酸−メタクリル酸メチルポリマーなどの腸溶剤基剤で被覆して腸溶剤製剤あるいはエチルセルロース、カルナウバロウ、硬化油などで被覆して持続性製剤とすることもできる。また、カプセル剤を製造するには、散剤又は顆粒剤を硬カプセルに充填するか、有効成分をそのまま或いはグリセリン、ポリエチレングリコール、ゴマ油、オリーブ油などに溶解した後ゼラチン膜で被覆し軟カプセルとすることができる。
【0048】
注射剤を製造するには、有効成分を必要に応じて塩酸、水酸化ナトリウム、乳糖、乳酸、ナトリウム、リン酸一水素ナトリウム、リン酸二水素ナトリウムなどのpH調整剤、塩化ナトリウム、ブドウ糖などの等張化剤と共に注射用蒸留水に溶解し、無菌濾過してアンプルに充填するか、更にマンニトール、デキストリン、シクロデキストリン、ゼラチンなどを加えて真空凍結乾燥し、用事溶解型の注射剤としても良い。また、有効成分にレチシン、ポリソルベート80、ポリオキシエチレン硬化ヒマシ油などを加えて水中で乳化せしめ注射剤用乳剤とすることもできる。
【0049】
直腸投与剤を製造するには、有効成分をカカオ脂、脂肪酸のトリ、ジ及びモノグリセリド、ポリエチレングリコールなどの座剤用基材と共に加湿して溶解し型に流し込んで冷却するか、有効成分をポリエチレングリコール、大豆油などに溶解した後、ゼラチン膜で被覆すれば良い。
【0050】
本発明の医薬の投与量及び投与回数は特に限定されず、治療対象疾患の悪化・進展の防止及び/又は治療の目的、疾患の種類、患者の体重や年齢、疾患の重篤度などの条件に応じて、医師の判断により適宜選択することが可能である。一般的には、経口投与における成人一日あたりの投与量は0.01〜1000mg(有効成分重量)程度であり、一日1回又は数回に分けて、或いは数日ごとに投与することができる。注射剤として用いる場合には、成人に対して一日量0.001〜100mg(有効成分重量)を連続投与又は間欠投与することが望ましい。
【0051】
本発明の医薬は、植込錠及びマイクロカプセルに封入された送達システムなどの徐放性製剤として、体内から即時に除去されることを防ぎ得る担体を用いて調製することができる。担体として、例えば、エチレンビニル酢酸塩、ポリ酸無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、及びポリ乳酸などの、生物分解性、生物適合性ポリマーを用いることができる。このような材料は、当業者によって容易に調製することができる。また、リポソームの懸濁液も薬剤的に受容可能な担体として使用することができる。有用なリポソームは、限定はしないが、ホスファチジルコリン、コレステロール及びPEG誘導ホスファチジルエタノール(PEG−PE)を含む脂質組成物として、使用に適するサイズになるように、適当なポアサイズのフィルターを通して調製され、逆相蒸発法によって精製される。
【0052】
本発明の医薬は、医薬組成物としてキットの形態で、容器、パック中に投与の説明書と共に含めることができる。本発明の医薬組成物がキットとして供給される場合、該組成物のうち異なる構成成分が別々の容器中に包装され、使用直前に混合される。このように構成成分を別々に包装するのは、活性構成成分の機能を失うことなく長期間の貯蔵を可能にするためである。
キット中に含まれる試薬は、構成成分の活性を長期間有効に持続し、容器内側に吸着することなく、また、構成成分を変質することのない材質で製造された容器中に供給される。例えば、封着されたガラスアンプルは、窒素ガスのような中性で不反応性を示すガスの存在下で封入されたバッファーなどを含んでも良い。アンプルは、ガラス、ポリカーボネート、ポリスチレンなどの有機ポリマー、セラミック、金属、又は試薬を保持するために通常用いられる他の何れかの適切な材料によって構成される。
【0053】
また、キットには使用説明書が添付されても良い。本キットの使用説明は、紙などに印刷され、及び/又はフロッピー(登録商標)ディスク、CD−ROM、DVD−ROM、Zipディスク、ビデオテープ、オーディオテープなどの電気的又は電磁的に読み取り可能な媒体に保存されて使用者に供給されても良い。詳細な使用説明は、キット内に実際に添付されていてもよく、あるいは、キットの製造者又は分配者によって指定され又は電子メール等で通知されるウェブサイトに掲載されていても良い。
【0054】
さらに、本発明には、PPARγ転写活性の異常に起因して発症する疾患等(例えば、癌の他、糖尿病、高脂血症、高コレステロール症、高血圧症、肥満症、動脈硬化症など)の予防又は治療方法が含まれる。
ここで「治療」とは、PPARγ転写活性の異常に起因して発症する疾患等に罹患した哺乳動物において、その病態の進行及び悪化を阻止又は緩和することを意味し、これによって該疾患の進行及び悪化を阻止又は緩和することを目的とする処置のことである。
また、「予防」とは、PPARγ転写活性の異常に起因して発症する疾患等に罹患するおそれがある哺乳動物について、該疾患の発症又は罹患を予め阻止することを意味し、これによって該疾患の諸症状等の発症を予め阻止することを目的とする処置のことである。
【0055】
治療の対象となる「哺乳動物」は、哺乳類に分類される任意の動物を意味し、特に限定はしないが、例えば、ヒトの他、イヌ、ネコ、ウサギなどのペット動物、ウシ、ブタ、ヒツジ、ウマなどの家畜動物などのことである。特に好ましい「哺乳動物」は、ヒトである。
【0056】
以下に実施例を示してさらに詳細に説明するが、本発明は実施例により何ら限定されるものではない。
【実施例】
【0057】
〔実施例1〕(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸〔化24〕の合成例
【化24】
式(I)の化合物の1つである(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸を以下の通り合成した。
【0058】
ベンジル 5−((R)−2−ベンジル−3−((S)−4−ベンジル−2−オキソオキサゾリジン−3−イル)−3−オキソプロピル)−2−プロポキシベンゾアートの合成
【化25】
(4S)−4−ベンジル−3−(3−フェニルプロパノイル)オキサゾリジン−2−オン(0.74g,2.37mmol)と脱水THF(20ml)をアルゴン雰囲気下混合した。反応液を−50℃に冷却し、LiHMDSのTHF溶液(8.0ml,8.0mmol)をシリンジで注加した。同温度で20分撹拌後、−15℃に昇温後さらに25分撹拌した。再度−50℃に冷却し、ベンジル 5−(ブロモメチル)−2−プロポキシ−ベンゾアート(0.86g,2.37mmol)を脱水THF(10ml)に溶かした溶液をゆっくり滴下した。反応液は75分かけて段階的に−50℃から0℃に昇温させた。0℃ にてさらに1時間撹拌後10%塩化アンモニウム水溶液中に反応液を注加し、酢酸エチル抽出(5×30ml)した。 抽出液は飽和食塩水で洗浄後無水硫酸マグネシウム乾燥し、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 9:2 v/v)、黄色油状の目的化合物を0.89g、収率64%で得た。
MS(FAB,(M+H)+) m/z 592 C37H37NO6
【0059】
5−((R)−2−ベンジル−3−((S)−4−ベンジル−2−オキソオキサゾリジン−3−イル)−3−オキソプロピル)−2−プロポキシ安息香酸の合成
【化26】
10% Pd/C (0.20g)、上記ベンジル 5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソプロピル]−2−プロポキシベンゾアート(0.89g,1.50mmol)及びAcOEt(80ml)を混合した。反応液を室温にて2.25時間接触還元を行った。反応液をセライトろ過し、ろ液を濃縮し、酢酸エチルで洗浄後ろ液と洗浄液を合わせ濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 5:2 v/v)、無色油状の目的化合物を0.41g、収率55%で得た。
1H NMR(400MHz,CDCl3)δ10.98(s,1H),8.06(d,J=2.4Hz,1H),7.52(dd,J=8.4,2.4Hz,1H),7.24−7.22(m,5H),7.20−7.16(m,3H),7.03(dd,J=7.2,2.0Hz,2H),6.97(d,J=8.4Hz,1H),4.56−4.48(m,1H),4.46−4.40(m,1H),4.22−4.13(m,2H),3.97(dd,J=8.8,2.4Hz,2H),3.81(t,J=8.2Hz,1H),3.16(dd,J=13.8,8.2Hz,1H),3.08−2.98(m,2H),2.84−2.76(m,2H),2.54(dd,J=13.4,9.4Hz,1H),1.96−1.88(m,2H),1.08(t,J=7.2Hz,3H);
MS(FAB,(M+H)+) m/z 502 C30H31NO6.
【0060】
(S)−4−ベンジル−3−((R)−2−ベンジル−3−(3−(ヒドロキシメチル)−4−プロポキシフェニル)プロパノイル)オキサゾリジン−2−オンの合成
【化27】
上記5−[(R)−2−ベンジルl−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシ安息香酸(0.41g,0.82mmol)と脱水THF(30ml)を混合した。アルゴン雰囲気下反応液を0℃に冷却し、1mol/LのBH3−THFコンプレックス1.60mL(1.60mmol)をシリンジで滴下した。反応液は0℃で2時間撹拌後室温にて一晩撹拌した。反応液を飽和塩化アンモニウム水溶液中に注加し、酢酸エチルにて抽出した。抽出液は飽和食塩水洗浄後無水硫酸マグネシウム乾燥し濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 3:1 v/v)、無色油状の目的化合物を0.40g、収率100%で得た。
1H NMR(400MHz,CDCl3)δ7.28−7.15(m,10H),6.99(dd,J=7.2,2.0Hz,2H),6.78(d,J=8.4Hz,1H),4.66(d,J=6.4Hz,2H),4.60−4.53(m,1H),4.43−4.38(m,1H),3.98−3.89(m,2H),3.77(t,J=8.2Hz,1H),3.08(dd,J=13.4,8.6Hz,1H),3.01−2.94(m,2H),2.85−2.75(m.2H),2.49(dd,J=13.4,9.4Hz,1H),2.37(t,J=6.6Hz,1H),1.85−1.76(m,2H),1.03(t,J=7.2Hz,3H);
MS(FAB,(M+H)+) m/z 488 C30H33NO5.
【0061】
5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシベンズアルデヒドの合成
【化28】
活性化二酸化マンガン(5.7g,65.6mmol)、上記(S)−4−ベンジル−3−[(R)−2−ベンジル−3−[3−(ヒドロキシメチル)−4−プロポキシ−フェニル]プロパノイル]オキサゾリジン−2−オン(0.40g,3.0mmol)とCH2Cl2(40ml)を混合し23時間還流した。反応液をアセトンで希釈しセライトを通して反応液を濾過し、ろ液を濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 3:1 v/v)、無色油状の目的化合物を0.20g、収率50%で得た。
1H NMR(400MHz,CDCl3)δ10.47(d,J=2.8Hz,1H),7.70(d,J=2.0Hz,1H),7.49(dd,J=8.4,2.4Hz,1H),7.27−7.16(m,8H),7.01(dd,J=7.4,1.8Hz,2H),6.90(d,J=8.8Hz,1H),4.56−4.48(m,1H),4.45−4.39(m,1H),4.03−3.97(m,2H),3.95(dd,J=8.8,2.4Hz,1H),3.80(t,J=8.4Hz,1H),3.12(dd,J=13.6,8.8Hz,1H),3.05−2.97(m,2H),2.79(dd,J=14.0,6.4Hz,2H),2.51(dd,J=13.2,9.2Hz,1H),1.89−1.80(m,2H),1.05(t,J=7.4Hz,3H);
MS(FAB,(M+H)+) m/z 486 C30H31NO5.
【0062】
4−(1−アダマンチル)−N−[[5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシフェニル]メチル]ベンズアミドの合成
【化29】
4−(1−アダマンチル)ベンズアミド(0.27g,1.03mmol)、上記5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシ−ベンズアルデヒド(0.20g,0.41mmol)、トリエチルシラン(0.16ml,1.03mmol)、TFA(0.08ml,1.03mmol)および脱水トルエン(30ml)を混合し、アルゴン雰囲気下49時間加熱還流した。水(100ml)に反応液を注加し、酢酸エチルで抽出した。抽出液は飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、ろ過後濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 2:1 v/v)、褐色油状の目的化合物を0.29g、収率97%で得た。
1H NMR(300MHz,CDCl3)δ7.61(d,J=8.7Hz,2H),7.35(d,J=8.7Hz,2H),7.28−7.16(m,10H),6.88−6.85(m,2H),6.80(d,J=8.4Hz,1H),6.71(t,J=5.7Hz,1H),4.70−4.52(m,3H),4.42−4.34(m,1H),3.98−3.88(m,2H),3.74(t,J=8.4Hz,1H),3.07(dd,J=13.8,8.7Hz,1H),3.01−2.84(m,2H),2.79(dd,J=13.8,6.3Hz,2H),2.48(dd,J=13.2,9.0Hz,1H),2.10(s,3H),1.89−1.72(m,14H),1.03(t,J=7.5Hz,3H);
MS(FAB,(M+H)+) m/z 725 C47H52N2O5.
【0063】
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸(実施例1の目的化合物)の最終合成過程
30 % H2O2(0.4 mL,4.0mmol)、上記合成の4−(1−アダマンチル)−N−[[5−[(R)−2−ベンジル−3−[(S)−4−ベンジル−2−オキソ−オキサゾリジン−3−イル]−3−オキソ−プロピル]−2−プロポキシフェニル]メチル]ベンズアミド(290mg,0.4mmol)、THF(24ml)、水(6ml)を混合し、次いで、リチウムヒドロキシド モノヒドラート(100mg,2.38mmol)を水(2ml)に溶かし加えた。アルゴン雰囲気下、反応液を0℃にて、2.5時間、室温で3時間撹拌後NaHSO3(1.0g)をH2O(6ml)に溶かし反応液に加えた。反応液を10%塩酸で希釈し酢酸エチルで抽出した。抽出液は飽和食塩水で洗浄し、無水硫酸マグネシウム乾燥後、ろ過、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 2:1 v/v)、無色粉末状の目的化合物(実施例1の目的化合物)を0.19g、収率86%で得た。
mp 177−178℃.
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.1Hz,2H),7.39(d,J=8.4Hz,2H),7.26−7.14(m,6H),7.03(dd,J=8.1,1.8Hz,1H),6.77−6.70(m,2H),4.64−4.51(m,2H),3.94(t,J=6.5Hz,2H),3.01−2.71(m,5H),2.10(s,3H),1.90−1.72(m,14H),1.06(t,J=7.4Hz,3H);
MS(FAB,(M+H)+)m/z 566 C37H43NO4 .
[α]D −3°(c0.10,CH3CN,26℃).
【0064】
〔実施例2〕(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸〔化30〕の合成例
【化30】
実施例1と同様の操作により、実施例2の化合物を合成した。
mp 177−178℃.
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.1Hz,2H),7.39(d,J=8.1Hz,2H),7.26−7.14(m,6H),7.03(dd,J=8.1,1.8Hz,1H),6.77−6.70(m,2H),4.64−4.51(m,2H),3.94(t,J=6.5Hz,2H),3.01−2.71(m,5H),2.10(s,3H),1.90−1.72(m,14H),1.06(t,J=7.4Hz,3H);
MS(FAB,(M+H)+)m/z 566 C37H43NO4.
[α]D +3°(c0.10,CH3CN,26℃).
【0065】
〔実施例3〕(S)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸〔化31〕の合成例
【化31】
実施例1と同様の操作により、実施例3の化合物を合成した。
mp 140−141℃.
1H NMR(300MHz,CDCl3)δ7.69(d,J=8.4Hz,2H),7.39(d,J=8.1Hz,2H),7.26−7.13(m,6H),7.02(dd,J=8.4,2.1Hz,1H),6.76−6.70(m,2H),4.59(d,J=4.5Hz,2H),3.93(t,J=6.45Hz,2H),2.91(dd,J=13.1,7.7Hz,1H),2.77−2.53(m,4H),2.10(s,3H),2.04−1.72(m,17H),1.05(t,J=7.4Hz,3H);
MS(FAB,(M+H)+) m/z 580 C38H45NO4.
[α]D −4°(c0.25,CH3CN,22℃).
【0066】
〔実施例4〕(R)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸の〔化32〕合成例
【化32】
実施例1と同様の操作により、実施例4の化合物を合成した。
mp 140−141℃.
1H NMR(300MHz,CDCl3)δ7.69(d,J=8.4Hz,2H),7.39(d,J=8.1Hz,2H),7.26−7.13(m,6H),7.02(dd,J=8.4,2.1Hz,1H),6.76−6.69(m,2H),4.59(d,J=4.5Hz,2H),3.93(t,J=6.5Hz,2H),2.91(dd,J=13.1,7.7Hz,1H),2.77−2.53(m,4H),2.10(s,3H),2.04−1.72(m,17H),1.05(t,J=7.4Hz,3H);
MS(FAB,(M+H)+) m/z 580 C38H45NO4.
[α]D+4°(c0.25,CHCl3,22℃).
【0067】
〔実施例5〕(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸〔化33〕の合成例
【化33】
実施例1と同様の操作により、実施例5の化合物を合成した。
mp 177−178℃.
1H NMR(300MHz,CDCl3)δ7.70(d,J=8.4Hz,2H),7.39(d,J=8.4Hz,2H),7.14(s,1H),7.04(dd,J=8.4,2.1Hz,1H),6.78−6.70(m,2H),4.65−4.53(m,2H),3.95(t,J=6.5Hz,2H),2.87−2.64(m,3H),2.10(s,3H),1.90−1.54(m,20H),1.34−1.12(m,5H),1.06(t,J=7.4Hz,3H),0.92−0.73(m,2H);
MS(FAB,(M+H)+) m/z 572 C37H49NO4.
[α]D −5.5°(c0.50,CHCl3,23℃).
【0068】
〔実施例6〕(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸〔化34〕の合成例
【化34】
実施例1と同様の操作により、実施例6の化合物を合成した。
mp 177−178℃.
1H NMR(300MHz,CDCl3)δ7.70(d,J=8.4Hz,2H),7.39(d,J=8.4Hz,2H),7.14(s,1H),7.03(dd,J=8.4,2.4Hz,1H),6.78−6.70(m,2H),4.65−4.53(m,2H),3.95(t,J=6.5Hz,2H),2.87−2.64(m,3H),2.10(s,3H),1.90−1.54(m,20H),1.34−1.12(m,5H),1.06(t,J=7.4Hz,3H),0.92−0.73(m,2H);
MS(FAB,(M+H)+) m/z 572 C37H49NO4.
[α]D +5.5°(c0.50,CHCl3,21℃).
【0069】
〔実施例7〕(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸〔化35〕の合成例
【化35】
式(I)の化合物の1つである(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸〔化35〕の合成例を以下に示す。
式(I)を合成するための中間化合物である式(II)の化合物の1つである4−アダマンチル−2−フルオロ−N−(4−フルオロ−5−ホルミル−2−プロポキシベンジル)ベンズアミド〔化36〕を以下のように合成した。
【化36】
【0070】
3−フルオロ−1−プロポキシベンゼンの合成
【化37】
3−フルオロフェノール(2.00g,17.8mmol)とDMF(6ml)を混合し、1−ヨードプロパン(4.25g,25.0mmol)および無水炭酸カリウム、K2CO3(2.76g,20.0mmol)を加え、室温にて一晩撹拌した。反応液は飽和塩化アンモニウム水溶液中に注加し、塩化メチレンで抽出した。抽出液は飽和食塩水で洗浄し、無水硫酸マグネシウム乾燥後、ろ過、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン)、無色油状の目的化合物を2.49g、収率91%で得た。
1H−NMR(500MHz,CDCl3)δ7.20(q,J=7.7Hz,1H),6.69−6.59(m,3H),3.90(t,J=6.7Hz,2H),1.81(m, 2H),1.04(t,J=7.6Hz,3H)
【0071】
4−フルオロ−2−プロポキシベンズアルデヒドの合成
【化38】
上記3−フルオロ−1−プロポキシベンゼン(2.39g,15.5mmol)とCH2Cl2(100mL)を混合し−78℃に冷却した。次にTiCl4(5mL)、CHCl2OCH3(2mL)を順次加え同温度で30分撹拌した。室温に戻しさらに一晩撹拌した。反応液は2mol/L塩酸中に注加し、塩化メチレンにて抽出した。抽出液は飽和食塩水で洗浄し、無水硫酸マグネシウム乾燥後、ろ過、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 5:1 v/v)、無色油状の目的化合物を1.74g、収率62%で得た。
1H−NMR(500MHz,CDCl3)δ10.39(s,1H),7.83(t,J=7.9Hz,1H),6.70−6.64(m,2H),4.00(t,J=6.4Hz,2H),1.87(m,2H),1.06(t,J=7.6Hz,3H)
【0072】
4−アダマンチル−2−フルオロ−N−(4−フルオロ−2−プロポキシベンジル)ベンズアミドの合成
【化39】
上記4−フルオロ−2−プロポキシベンズアルデヒド(661mg,3.63mmol)、2−フルオロ−4−アダマンチルベンズアミド(〔化18〕)(1.02g,3.73mmol)、トルエン(0.5mL)を混合し、トリフルオロ酢酸(0.5mL)、トリエチルシラン(1.6mL)を順次加えた。アルゴン雰囲気下、80℃にて3日撹拌した。反応液を濃縮し、残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 10/1から5/1 v/v)、無色油状の目的化合物を1.05g、収率66%で得た。
1H−NMR(500MHz,CDCl3)δ8.04(t,J=8.5Hz,1H),7.30−7.23(m,3H),7.05(d,J=14.6Hz,1H),6.61−6.58(m,2H),4.61(d,J=5.5Hz,2H),3.95(t,J=6.4 Hz,2H),2.10(s,3H),1.90−1.72(m,14H),1.08(t,J=7.3Hz,3H);
MS (FAB) 440 (M+H)+
【0073】
次に、上記4−アダマンチル−2−フルオロ−N−(4−フルオロ−2−プロポキシベンジル)ベンズアミド(1.01g,2.30mmol)と、CH2Cl2(20mL)を混合し−78℃に冷却した。次にTiCl4(1.0mL)、CHCl2OCH3(0.3mL)を順次加え同温度で30分撹拌した。室温に戻しさらに3時間撹拌した。反応液は2mol/L塩酸中に注加し、塩化メチレンにて抽出した。抽出液は飽和食塩水で洗浄し、無水硫酸マグネシウム乾燥後、ろ過、濃縮した。残留物はシリカゲルカラムクロマトグラフィーにて精製し(n−ヘキサン/AcOEt 5:1 v/v)、無色粉末の目的化合物(〔化36〕の化合物)を1.02g、収率62%で得た。
1H−NMR(500MHz,CDCl3)δ10.08(s,1H),8.04(t,J=8.5Hz,1H),7.83(d,J=7.9Hz,1H),7.26−7.21(m,2H),7.07(dd,J=14.6,1.8Hz,1H),6.62(d,J=12.2Hz,1H),4.64(d,J=5.5Hz,2H),4.04(t,J=6.7Hz,2H),2.11(s,3H),1.93−1.73(m,14H),1.09(t,J=7.3Hz,3H);
MS (FAB) 468 (M+H)+
【0074】
〔化36〕の合成と同様にして、式(II)の化合物の1つである4−アダマンチル−N−(5−ホルミル−2−プロポキシベンジル)ベンズアミド(〔化40〕の化合物)の合成も行った。
【化40】
1H NMR(300MHz,CDCl3)δ9.87(s,1H),7.86(d,J=2.1Hz,1H),7.82(dd,J=8.4,2.1Hz,1H),7.73(d,J=8.4Hz,2H),7.43(d,J=8.1Hz,1H),6.99(d,J=8.4Hz,1H),6.59(t,J=5.7Hz,1H),4.70(d,J=6.0Hz,2H),4.09(t,J=6.5Hz,2H),2.11(s,3H),1.90−1.73(m,14H),1.10(t,J=7.4Hz,3H)
MS(FAB,(M+H)+) m/z 432 C28H33NO3
【0075】
メチル 2−(ジメトキシホスホリル)−3−(2−フルオロフェニル)プロパノアートの合成
【化41】
カリウムtert−ブトキシド(0.36g,3.2mmol)の脱水THF(20mL)溶液に0℃、アルゴン雰囲気下、ホスホノ酢酸トリメチル(0.43mL,2.7 mmol)を加え30分撹拌した。次に2−フルオロベンジルブロミド(0.32mL,2.7mmol)の脱水THF溶液(10mL)を加えた後、室温で19時間撹拌した。反応液を水(50mL)中にあけ、酢酸エチル(30ml×5)で抽出した。有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥後濃縮した。残留物をシリカゲルカラムクロマトグラフィー(溶出液 n−ヘキサン:酢酸エチル=1:1 v/v)で精製し、0.66g(86%)の表題化合物を無色油状物質として得た。
MS(FAB,(M+H)+) m/z 291 C12H16FO5P
【0076】
メチル 2−(ジメトキシホスホリル)−3−(3−フルオロフェニル)プロパノアートの合成
【化42】
〔化41〕の化合物と同様にして、表題化合物を得た。
MS(FAB,(M+H)+) m/z 291 C12H16FO5P
【0077】
メチル 2−(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)アクリラートの合成
【化43】
カリウムtert−ブトキシド(0.15g,1.34mmol)の脱水THF(20mL)溶液に0℃、アルゴン雰囲気下、化合物メチル 2−(ジメトキシホスホリル)−3−(2−フルオロフェニル)プロパノアート(〔化41〕の化合物)(0.30g,1.03mmol)の脱水THF(4mL)溶液を加え、30分撹拌した。次にアルデヒド誘導体(〔化40〕の化合物)(0.45g,1.03mmol)の脱水THF溶液(4mL)を加えた後、室温で16時間撹拌した。反応液を水(50mL)中にあけ、酢酸エチル(30ml×5)で抽出した。有機層を飽和食塩水で、洗浄後無水硫酸マグネシウムで乾燥後濃縮した。残留物をシリカゲルカラムクロマトグラフィー(溶出液 n−ヘキサン:酢酸エチル=4:1 v/v)で精製し、0.42g(69%)の表題化合物を無色油状物質として得た。
MS(FAB,(M+H)+) m/z 597 C38H44FNO4
【0078】
メチル (2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパノアートの合成
【化44】
メチル 2−(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)アクリラート(0.42g,0.70mmol) の酢酸エチル(30mL)溶液に、10%パラジウム担持活性炭(50mg)を加え、室温、200−300kPa水素加圧化2時間水素添加した。触媒をセライトで濾取し、酢酸エチルで洗浄した。濾液を濃縮し、残留物をシリカゲルカラムクロマトグラフィー(溶出液、n−ヘキサン:酢酸エチル=4:1 v/v)で精製し、0.37g(89%)の表題化合物を無色油状物質として得た。
1H NMR(300MHz,CDCl3)=δ7.70(d,J=8.7Hz,2H),7.40(d,J=8.7Hz,2H),7.24−7.10(m,3H),7.05−6.95(m,3H),4.61(d,J=5.7Hz,2H),3.96(t,J=6.3Hz,2H),3.50(s,3H),3.00−2.89(m,4H),2,74(dd,J=13.2,4.8Hz,1H),2.10(s,3H),1.91−1.70(m,14H),1.06(t,J=7.2Hz,3H)
MS (FAB,(M+H)+) m/z 598 C38H44FNO4
【0079】
(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸(〔化35〕の化合物)の最終合成過程
上記合成のメチル (2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパノアート(0.37g,0.62mmol) のエタノール溶液に、2mol/L水酸化ナトリウム水溶液(3.1mL,6.2mmol)を加え60℃で48時間撹拌した。反応液を濃縮し10%塩酸を溶液がpH1になるまで加え撹拌後析出した固体を吸引濾過した。得られた固体を酢酸エチルで溶かし無水硫酸マグネシウムで乾燥した後濃縮した。残留物をシリカゲルカラムクロマトグラフィー (溶出液 n−ヘキサン:酢酸エチル=2:1 v/v)で精製し、0.32g(89%) の表題化合物(〔実施例7〕の目的化合物)を白色固体として得た。
m.p.158−159℃
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.7Hz,2H),7.39(d,J=8.4Hz,2H),7.21−7.15(m,3H),7.06−6.95(m,3H),6.77−6.72(m,2H),4.57(d,J=5.4Hz,2H),3.94(t,J=6.5Hz,2H),2.98−2.80(m,4H),2.76(dd,J=13.8,4.8Hz,1H),2.10(s,3H),1.90−1.71(m,14H),1.06(t,J=7.4Hz,3H)
MS (FAB, M+H)+) m/z 584 C37H42NO4F
【0080】
〔実施例8〕(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸〔化45〕の合成例
【化45】
【0081】
メチル 2−(3−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)アクリラートの合成
【化46】
〔化43〕の合成操作と同様にして、表題化合物を得た。
MS(FAB,(M+H)+) m/z 597 C38H44FNO4
【0082】
メチル (3−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパノアートの合成
【化47】
〔化44〕の合成操作と同様にして、表題化合物を無色油状物質として得た。
1H NMR(300MHz,CDCl3)δ7.70(d,J=8.7Hz,2H),7.40(d,J=8.7Hz,2H),7.24−7.17(m,1H),7.10(s,1H),7.01(d,J=8.4Hz,1H),6.92−6.76(m,4H),6.66(t,J=5.7Hz,1H),4.61(d,J=5.7Hz,2H),3.96(t,J=6.5Hz,2H),3.51(s,3H),2.93−2.86(m,3H),2.80−2.69(m,2H),2.10(s,3H),1.90−1.72(m,14H),1.07(t,J=7.5Hz,3H).
MS (FAB,(M+H)+) m/z 598 C38H44FNO4
【0083】
次に、実施例7の化合物と同様にして、実施例8の化合物(〔化45〕)を得た。
m.p.201−202℃
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.4Hz,2H),7.39(d,J=8.1Hz,2H),7.23−7.15(m,2H),7.02(dd,J=2.1,8.1Hz,1H),6.94−6.85(m,3H),6.78−6.71(m,2H),4.58(d,J=5.4Hz,2H),3.95(t,J=6.6Hz,2H),2.99−2.87(m,3H),2.79−2.72(m,2H),2.09(s,3H),1.90−1.71(m,14H),1.06(t,J=7.35Hz,3H).
MS (FAB,(M+H)+) m/z 584 C37H42FNO4
【0084】
〔実施例9〕(4−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸(〔化48〕の化合物)の合成例
【化48】
【0085】
メチル−(4−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパノアートの合成
【化49】
〔化47〕の合成操作と同様にして、表題化合物を無色油状物質として得た。
1H NMR(300MHz,CDCl3)δ7.70(d,J=8.4Hz,2H),7.40(d,J=8.4Hz,2H),7.12−7.06(m,3H),7.00(dd,J=8.3,2.3Hz,1H),6.93(t,J=8.7Hz,2H),6.77(d,J=8.4Hz,1H),6.66(t,J=5.6Hz,1H),4.61(d,J=5.7Hz,2H),3.96(t,J=6.5Hz,2H),3.49(s,3H),2.93−2.84(m,3H),2.78−2.66(m,2H),2.10(s,3H),1.91−1.72(m,14H),1.06(t,J=7.4Hz,3H).
MS (FAB,(M+H)+) m/z 598 C38H44FNO4
【0086】
次に、実施例7の化合物と同様にして、実施例9の化合物(〔化48〕)を得た。
m.p.167−168℃
1H NMR(300MHz,CDCl3)δ7.68(d,J=8.4Hz,2H),7.39(d,J=8.4Hz,2H),7.16−7.09(m,3H),7.02(dd,J=8.4,2.1Hz,1H),6.92(t,J=8.4Hz,2H),6.77−6.73(m,2H),4.58(d,J=6.0Hz,2H),3.95(t,J=6.5Hz 2H),2.96−2.85(m,3H),2.76−2.68(m,2H),2.10(s,3H),1.90−1.71(m,14H),1.06(t,J=7.4Hz,3H).
MS (FAB,(M+H)+) m/z 584 C37H42NO4F
【0087】
〔実施例10〕2−ベンジル−3−(2−フルオロ−5−((2−フルオロ−4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化50】
実施例7と同様の操作により、実施例10の化合物(〔化50〕)を得た。
1H−NMR(500MHz,CDCl3)δ8.00(t,J=8.5Hz,1H),7.35(m,1H),7.25−7.15(m,7H),7.05(dd,J=14.6,1.8Hz,1H),6.54(d,J=11.6Hz,1H),4.55(m,2H),3.91(t,J=6.4Hz,2H),3.02−2.96(m,2H),2.86−2.77(m,3H),2.10(s,3H),1.87−1.72(m,14H), 1.06(t,J=7.3Hz,3H);
MS (FAB) 602 (M+H)+
【0088】
〔実施例11〕2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸の合成例
【化51】
実施例7の同様の操作により、実施例11の化合物(〔化51〕)を得た。
1H−NMR(500MHz,CDCl3)δ8.02(t,J=8.5Hz,1H),7.34(m,1H),7.24−7.14(m,7H),7.04(dd,J=14.6,1.8Hz,1H),6.54(d,J=12.2Hz,1H),4.55(s,2H),3.91(t,J=6.7Hz,2H),2.86(m,2H),2.71(m,2H), 2.60(m,1H),2.09(s,3H),2.00−1.72(m,16H),1.06(t,J=7.3Hz,3H);
MS (FAB) 616 (M+H)+
【0089】
〔実施例12〕2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸の合成例
【化52】
実施例7と同様の操作により、実施例12の化合物(〔化52〕)を得た。
1H−NMR(500MHz,CDCl3)δ8.02(t,J=8.5Hz,1H),7.33(m,1H),7.28−7.12(m,7H),7.04(dd,J=14.6,1.8Hz,1H),6.54(d,J=11.6Hz,1H),4.55(d,J=5.5Hz,2H),3.91(t,J=6.7Hz,2H),2.85−2.77(m,2H),2.67−2.57(m,3H),2.10(s,3H),1.92−1.54(m,18H),1.07(t,J=7.3Hz,3H);
MS (FAB) 630 (M+H)+
【0090】
〔実施例13〕(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(ピリミジン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸の合成例
【化53】
実施例1と同様の操作により、実施例13の化合物〔化53〕を得た。
1H−NMR(400MHz,DMSO−d6)δ12.06(s,1H),8.94(d,J=4.8Hz,2H),8.91(t,J=5.8Hz,1H),8.47(d,J=8.8Hz,2H),8.04(d,J=8.4Hz,2H),7.49(t,J=4.8Hz,1H),7.19−7.16(m,2H),7.12−7.09(m,3H),7.02−7.01(m,2H),6.87(d,J=9.2Hz,1H),4.50−4.40(m,2H),3.93(t,J=6.4Hz,2H),2.82−2.71(m,3H),2.70−2.58(m,2H),1.77−1.69(m,2H),0.98(t,J=7.4Hz,3H)
【0091】
〔実施例14〕(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(チオフェン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸の合成例
【化54】
実施例1と同様の操作により、実施例14の化合物〔化54〕を得た。
1H−NMR(400MHz,DMSO−d6)δ12.05(s,1H),8.81(t,J=5.6Hz,1H),7.94(t,J=8.8Hz,2H),7.76(d,J=8.4Hz,2H),7.64(dd,J=3.6,1.2Hz,1H),7.62(dd,J=5.2,1.2Hz,1H),7.19−7.15(m,3H),7.12−7.09(m,3H),7.02−6.99(m,2H),6.86(d,J=8.4Hz,1H),4.49−4.38(m,2H),3.92(t,J=6.2Hz,2H),2.81−2.71(m,3H),2.68−2.56(m,2H),1.77−1.68(m,2H),0.98(t,J=7.4Hz,3H)
【0092】
〔実施例15〕(R)−2−ベンジル−3−(3−((4−(ピペリジン−1−イル)ベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化55】
実施例1と同様の操作により、実施例15の化合物〔化55〕を得た。
1H−NMR(400MHz,DMSO−d6)δ12.05(s,1H),8.46(t,J=5.8Hz,1H),7.76(d,J=8.8Hz,2H),7.18−7.08(m,5H),6.99(dd,J=8.4,2.0Hz,1H),6.95−6.92(m,3H),6.84(d,J=8.4Hz,1H),4.44−4.33(m,2H),3.91(t,J=6.4Hz,2H),3.27(t,J=5.2Hz,4H),2.77−2.69(m,3H),2.65−2.52(m,2H),1.77−1.68(m,2H),1.57(s,6H),0.98(t,J=7.4Hz,3H)
【0093】
〔実施例16〕(R)−2−ベンジル−3−(3−((2’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化56】
実施例1と同様の操作により、実施例16の化合物〔化56〕を得た。
1H−NMR(400MHz,CDCl3)δ7.80(d,J=8.4Hz,2H),7.59(dd,J=8.4,1.6Hz,2H),7.43(dt,J=7.6,1.8Hz,1H),7.37−7.32(m,1H),7.28−7.14(m,8H),6.80−6.76(m,2H),4.65−4.56(m,2H),3.95(t,J=6.6Hz,2H),3.01−2.87(m,3H),2.82−2.73(m,2H),1.88−1.79(m,2H),1.06(t,J=7.4Hz,3H)
【0094】
〔実施例17〕 (R)−2−ベンジル−3−(3−((3’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化57】
実施例1と同様の操作により、実施例17の化合物〔化57〕を得た。
1H−NMR(400MHz,DMSO−d6)δ8.87(t,J=5.6Hz,1H),8.00(d,J=8.4,2H),7.82(d,J=8.8Hz,2H),7.61−7.58(m,2H),7.55−7.49(m,1H),7.23(dt,J=8.8,1.6Hz,1H),7.19−7.15(m,2H),7.12−7.09(m,3H),7.03−7.00(m,2H),6.86(d,J=8.4Hz,1H),4.50−4.40(m,2H),3.92(t,J=6.4Hz,2H),2.79−2.71(m,3H),2.69−2.56(m,2H),1.77−1.69(m,2H),0.99(t,J=7.4Hz,3H)
【0095】
〔実施例18〕(R)−2−ベンジル−3−(3−((2’6’フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化58】
実施例1と同様の操作により、実施例18の化合物〔化58〕を得た。
1H−NMR(400MHz,DMSO−d6)δ12.06(s,1H),7.95(d,J=8.4Hz,2H),7.57(d,J=8.4,2H),7.53−7.46(m,1H),7.25(t,J=8.2Hz,2H),7.17−7.14(m,2H),7.11−7.09(m,3H),7.03−7.00(m,2H),6.87(d,J=8.4Hz,1H),4.51−4.40(m,2H),3.93(t,J=6.4Hz,2H),2.81−2.69(m,3H),2.68−2.57(m,2H),1.78−1.69
(m,2H),0.99(t,J=7.4Hz,3H)
【0096】
〔実施例19〕 (R)−2−ベンジル−3−(3−(2’−メチルビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化59】
実施例1と同様の操作により、実施例19の化合物〔化59〕を得た。
1H−NMR(400MHz,CDCl3)δ7.78(d,J=8.4Hz,2H),7.35(d,J=8.4Hz,2H),7.28−7.22(m,5H),7.20−7.17(m,5H),7.04(dd,J=8.4,2.2Hz,1H),6.81−6.76(m,2H),4.65−4.56(m,2H),3.96(t,J=6.4Hz,2H),3.02−2.87(m,3H),2.82−2.73(m,2H),2.24(s,3H),1.88−1.80(m,2H),1.06(t,J=7.4Hz,3H)
【0097】
〔実施例20〕 (R)−2−ベンジル−3−(3−(ビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸の合成例
【化60】
実施例1と同様の操作により、実施例20の化合物〔化60〕を得た。
1H−NMR(400MHz,CDCl3)δ7.80(d,J=8.4Hz,2H),7.63−7.57(m,4H),7.45(t,J=7.4Hz,2H),7.37(t,J =7.4Hz,1H),7.27−7.23(m,2H),7.04(dd,J=8.4,2.4Hz,1H),6.81(t,J=5.8Hz,1H),6.77(d,J=8.0Hz,1H),4.65−4.55(m,2H),3.95(t,J=6.4Hz,2H),3.02−2.86(m,3H),2.80−2.73(m,2H),1.88−1.79(m,2H),1.05(t,J=7.4Hz,3H)
【0098】
〔実験例〕
1.本発明の化合物のPPARγアゴニスト活性の測定
10%脱脂牛血清を含むダルベッコ変法イ−グル培地(FCS/DMEM)にて培養したヒト胎児腎細胞(HEK293)に、酵母の転写因子(GAL4)のDNA結合領域とヒト型PPARの各サブタイプ(α、δ、γ)のリガンド結合領域との融合蛋白質を発現する受容体プラスミド及びそのレポータープラスミド、さらに内部標準用のβガラクトシダーゼプラスミドをリン酸カルシウム法にて無血清状態にてコトランスフェクションした。その後、被検化合物を添加して16時間後にルシフェラ−ゼ活性ならびにβガラクトシダーゼ活性を測定し、内部標準により補正した。
上記方法により測定したPPAR(実施例1〜12)に対するEC50の値を表1に示す。
【表1】
【0099】
また、一般式(I)のR1をアダマンチル基以外の置換基にして、水溶性の向上を試みた(実施例13〜20)。この結果を表2に示す。
【表2】
【0100】
表1に示すように、本発明の化合物は、PPARγに選択的に作用するアゴニスト活性を有する。これらのEC50は、同様の方法で測定した既知のPPARγ選択的なアゴニストであるロシグリタゾン(EC50=43nM)やPPARのパンアゴニストであるTIPP703((S)−2−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシベンジル)ブタン酸、EC50=37nM;Bioorg Med Chem Lett. 2008, 18(3), 1110-1115に記載の化合物番号12)と比較しても、同等以上の値を示しており、良好なPPARγ選択的アゴニストであることが明らかとなった。
また、一般式(I)のR1をアダマンチル基以外の置換基にすると、EC50は、若干悪くなるものもあるが、水溶性は、R1がアダマンチル基である化合物(例えば、実施例1の化合物)に比較して、改善されている。特に、実施例13、15及び18の化合物は、実施例1の化合物よりも水溶性が向上し、かつ、EC50の値についても、既知のPPARγ選択的なアゴニストであるロシグリタゾンと同等以上の値を示している。
【0101】
2.PPARγアゴニストの癌細胞への影響
次に、本発明の化合物が癌細胞に及ぼす影響について検討を行った。本実施例では、典型的なスキルス胃癌の臨床経過を呈するスキルス胃癌細胞株OCUM−2MD3(大阪市立大学医学部の八代正和先生から供与を受けた)を使用して実験を行った。また、正常細胞に対する化合物の影響をみるために、正常細胞株であるOUMS−24(Bai L, et al., Int J Cancer. 1993 ; 53: 451-456)を使用して実験を行った。
OCUM−2MD3又はOUMS−24を6ウェルプレートに1.5×104細胞/ウェル(培地:DMEM+10% FBS)となるように播き、翌日(Day1)、本発明の化合物(実施例1の化合物、実施例13の化合物及び実施例15の化合物)、トログリタゾン及びDMSOのみで処理を行い、4日目(Day4)に化合物を含む培地で培地交換を行い、5日目(Day5)に細胞数の計測を行った(5視野にて計測を行った)。また、アポトーシス細胞の割合は、(トリパンブルー染色細胞数)/(総細胞数)により算出した。
図1に実施例1の化合物によるアポトーシスの誘導効果及び細胞増殖抑制効果に関する計測結果を示す。実施例1の化合物を、0.1μM、1μM、10μM及び100μMとなるように添加し、コントロールとしてトログリタゾンを10μM及び100μMとなるように添加した。トログリタゾンが100μMで活性(アポトーシス誘導活性及び細胞増殖抑制活性)を示すのに対し、実施例1の化合物はその1/10である10μMでアポトーシス誘導効果及び細胞増殖抑制効果を示した。
【0102】
次に、癌細胞ではない正常な細胞に対し、本発明の化合物が与える影響について検討を行った。図2は、本発明の化合物(実施例1の化合物、実施例13の化合物及び実施例15の化合物)のスキルス胃癌細胞(OCUM−2MD)及び正常細胞(OUMS−24)に対するアポトーシス誘導効果について、検討を行った結果である。実施例1の化合物については、その濃度依存的にOCUM−2MDのアポトーシスを誘導したのに対し、OUMS−24については、少なくとも10μMまでは、アポトーシスを誘導しなかった。一方、実施例13及び実施例15の化合物については、100μMにおいても、OCUM−2MDに選択的にアポトーシスを誘導した。
以上の結果から、本発明の化合物は、トログリタゾンよりも低用量で癌細胞に対する増殖抑制効果を示し(図1)、かつ、正常細胞の増殖にはほとんど影響を与えない(図2)ことが分かる。
【産業上の利用可能性】
【0103】
本発明の化合物は、PPARγのアゴニストの活性を有する。従って、これらの化合物を有効成分として含有する医薬は、PPARγの活性異常に起因する疾患の治療等に効果を発揮することが期待される。
【特許請求の範囲】
【請求項1】
下記一般式(I)で表されるα−置換フェニルプロピオン酸誘導体若しくはその薬学上許容される塩又は水和物。
【化1】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、nは0から10の整数を表す]
【請求項2】
下記一般式(I)で表されるPPARγに選択的なアゴニスト。
【化2】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、nは0から10の整数を表す]
【請求項3】
R1がアダマンチル基、ピリミジニル基、ピペリジニル基、2−チエニル基、2−フルオロフェニル基、3−フルオロフェニル基、2,6−ジフルオロフェニル基のいずれかである請求項2に記載のPPARγに選択的なアゴニスト。
【請求項4】
R2が水素原子又はフッ素原子である請求項2に記載のPPARγに選択的なアゴニスト。
【請求項5】
R3がプロピル基である請求項2に記載のPPARγに選択的なアゴニスト。
【請求項6】
R4が水素原子又はフッ素原子である請求項2に記載のPPARγに選択的なアゴニスト。
【請求項7】
R5が無置換フェニル基又はフルオロフェニル基である請求項2に記載のPPARγに選択的なアゴニスト。
【請求項8】
一般式(I)で表される化合物が、
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸、
(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸、
(S)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸、
(R)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸、
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸、
(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸、
(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
(3−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
(4−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
2−ベンジル−3−(2−フルオロ−5−((2−フルオロ−4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸、
2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸、
(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(ピリミジン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸
(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(チオフェン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸
(R)− 2−ベンジル−3− (3−((4−(ピペリジン−1−イル)ベンズアミド)メチル)−4−プロポキシフェニル) プロパン酸、
(R)−2−ベンジル−3−(3−((2’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル) プロパン酸
(R)−2−ベンジル−3−(3−((3’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル) プロパン酸
(R)−2−ベンジル−3−(3−((2’6’ジフルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル) プロパン酸
(R)−2−ベンジル−3−(3−(2’−メチルビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−(ビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
のいずれかである請求項2に記載のPPARγに選択的なアゴニスト。
【請求項9】
請求項2乃至8のいずれかに記載のPPARγに選択的なアゴニストを含むPPARγ転写活性化剤。
【請求項10】
請求項2乃至8のいずれかに記載のPPARγに選択的なアゴニストを含む抗癌剤。
【請求項11】
下記一般式(II)で表される、一般式(I)の化合物合成のための中間体化合物。
【化3】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基を表す]
【請求項12】
下記一般式(III)で表される、一般式(I)の化合物合成のための中間体化合物。
【化4】
[式中、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R4は水素原子又はハロゲン原子、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、Pは、不斉補助基又は炭素数1から10の直鎖状又は分岐状アルキル基、nは0から10の整数を表す]
【請求項13】
一般式(II)又は一般式(III)で表される化合物を用いた、一般式(I)で表される化合物の製造方法。
【請求項1】
下記一般式(I)で表されるα−置換フェニルプロピオン酸誘導体若しくはその薬学上許容される塩又は水和物。
【化1】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、nは0から10の整数を表す]
【請求項2】
下記一般式(I)で表されるPPARγに選択的なアゴニスト。
【化2】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、nは0から10の整数を表す]
【請求項3】
R1がアダマンチル基、ピリミジニル基、ピペリジニル基、2−チエニル基、2−フルオロフェニル基、3−フルオロフェニル基、2,6−ジフルオロフェニル基のいずれかである請求項2に記載のPPARγに選択的なアゴニスト。
【請求項4】
R2が水素原子又はフッ素原子である請求項2に記載のPPARγに選択的なアゴニスト。
【請求項5】
R3がプロピル基である請求項2に記載のPPARγに選択的なアゴニスト。
【請求項6】
R4が水素原子又はフッ素原子である請求項2に記載のPPARγに選択的なアゴニスト。
【請求項7】
R5が無置換フェニル基又はフルオロフェニル基である請求項2に記載のPPARγに選択的なアゴニスト。
【請求項8】
一般式(I)で表される化合物が、
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸、
(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−フェニルプロパン酸、
(S)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸、
(R)−2−(4−プロポキシ−3−(((4−アダマンタンチル)ベンズアミド)メチル)ベンジル)−4−フェニルブタン酸、
(R)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸、
(S)−2−((3−(((4−(1−アダマンチル)ベンゾイル)アミノ)メチル)−4−プロポキシフェニル)メチル)−3−シクロヘキシルプロパン酸、
(2−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
(3−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
(4−フルオロベンジル)−3−(3−((4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
2−ベンジル−3−(2−フルオロ−5−((2−フルオロ−4−アダマンチルベンズアミド)メチル)−4−プロポキシフェニル)プロパン酸、
2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸、
2−(2−フルオロ−5−((4−アダマンチル−2−フルオロベンズアミド)メチル)−4−プロポキシベンジル)−4−フェニルブタン酸、
(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(ピリミジン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸
(R)−2−ベンジル−3−(4−プロポキシ−3−((4−(チオフェン−2−イル)ベンズアミド)メチル)フェニル)プロパン酸
(R)− 2−ベンジル−3− (3−((4−(ピペリジン−1−イル)ベンズアミド)メチル)−4−プロポキシフェニル) プロパン酸、
(R)−2−ベンジル−3−(3−((2’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル) プロパン酸
(R)−2−ベンジル−3−(3−((3’−フルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル) プロパン酸
(R)−2−ベンジル−3−(3−((2’6’ジフルオロビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル) プロパン酸
(R)−2−ベンジル−3−(3−(2’−メチルビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
(R)−2−ベンジル−3−(3−(ビフェニル−4−イルカルボキサミド)メチル)−4−プロポキシフェニル)プロパン酸
のいずれかである請求項2に記載のPPARγに選択的なアゴニスト。
【請求項9】
請求項2乃至8のいずれかに記載のPPARγに選択的なアゴニストを含むPPARγ転写活性化剤。
【請求項10】
請求項2乃至8のいずれかに記載のPPARγに選択的なアゴニストを含む抗癌剤。
【請求項11】
下記一般式(II)で表される、一般式(I)の化合物合成のための中間体化合物。
【化3】
[式中、R1はアダマンチル基、無置換又は置換基を有していても良いフェニル基、ピリジル基、ピリミジニル基、ピラジニル基、ピペリジン基、アゼピル基若しくはピペラジニル基(置換基は、ハロゲン原子)、あるいは、2−チエニル基、モルホリノ基、R2及びR4は同一又は相異なって水素原子又はハロゲン原子、R3は炭素数1から10の直鎖状又は分岐状アルキル基を表す]
【請求項12】
下記一般式(III)で表される、一般式(I)の化合物合成のための中間体化合物。
【化4】
[式中、R3は炭素数1から10の直鎖状又は分岐状アルキル基、R4は水素原子又はハロゲン原子、R5は置換基を有していても良いフェニル基又はシクロヘキシル基(置換基は、ハロゲン原子、炭素数1から10の直鎖状又は分岐状アルキル基、炭素数1から10の直鎖状又は分岐状アルコキシ基)、Pは、不斉補助基又は炭素数1から10の直鎖状又は分岐状アルキル基、nは0から10の整数を表す]
【請求項13】
一般式(II)又は一般式(III)で表される化合物を用いた、一般式(I)で表される化合物の製造方法。
【図1】


【図2】
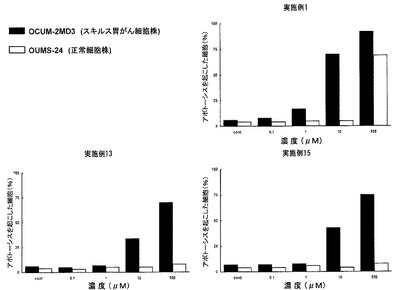
【図2】
【公開番号】特開2012−41334(P2012−41334A)
【公開日】平成24年3月1日(2012.3.1)
【国際特許分類】
【出願番号】特願2011−156216(P2011−156216)
【出願日】平成23年7月15日(2011.7.15)
【出願人】(504147243)国立大学法人 岡山大学 (444)
【Fターム(参考)】
【公開日】平成24年3月1日(2012.3.1)
【国際特許分類】
【出願日】平成23年7月15日(2011.7.15)
【出願人】(504147243)国立大学法人 岡山大学 (444)
【Fターム(参考)】
[ Back to top ]